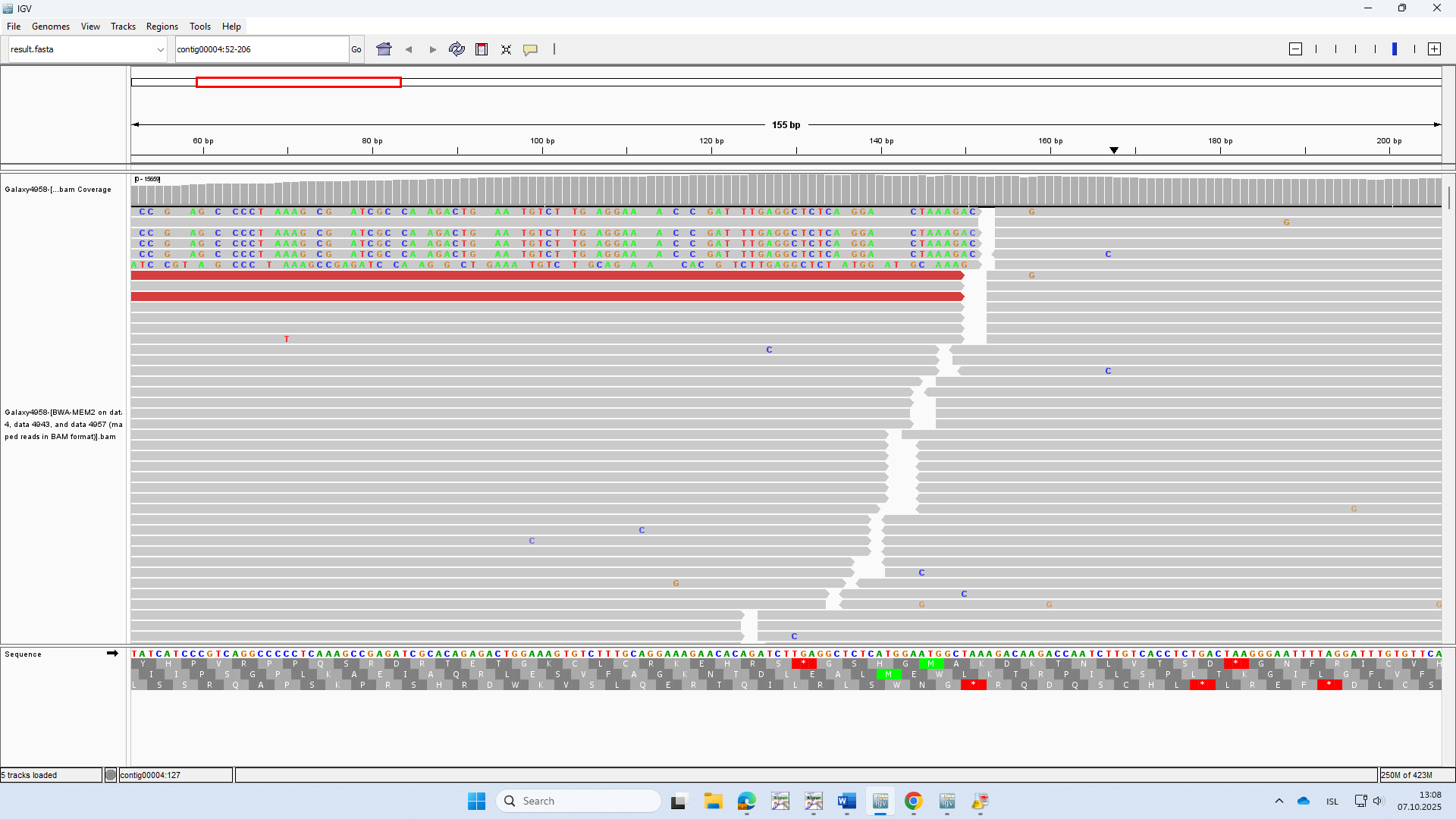
When using your BWA-MEM2 tool to align sequencing reads to a reference genome, I noticed apparent frameshift mutations in a few reads — not soft-clippings (see example below). These changes seem to result from the alignment process rather than genuine biological mutations.
Upon closer inspection, the affected reads appear to be slightly shifted by a few bases relative to the reference. Could you please advise how to correct or filter out such artifacts?
Best regards, Chen Huiping
University Hospital of Iceland
Then, to control for low quality alignments, you can Filter BAM reads out from the result. Removing reads that do not have the quality properties you are interested in retaining could involve only keeping primary alignments, or alignments from proper pairs, or alignments with a mapQ value of 20 or 30.
Then, depending on the protocol, you might want to Mark Duplicates or BamLeftAlign.
You can also tune parameters with the alignment tool itself. Most of the command line options are on the tool forms of the mapping tools and some have preset groupings for specific read types.
From what you have now:
Yes, you have an insertion that has shifted one of the reads in your group but none of those sequences appear to be good representatives of the reference sequence. Are you sure that all of this data was generated using the same exact reference sequence assembly? There seems to be more going on here. I would start by confirming the upstream steps. Did the mapping job use the same reference fasta as you are using in the IGV browser?
For a discussion about how different assembly versions can lead to issues, see this guide. It is focused on human but all genomes work the same way. Meaning, slight differences between versions of assemblies can lead to coordinate mismatch issues. → Reference genomes at public Galaxy servers: GRCh38/hg38 example
The red alignments were not aligned by BWA-MEM2, given any other filtering criteria that you may have set. You can reset to the defaults if you need to start over.
But, if you think those regions should be aligned instead, you will have to go back to the mapping step and adjust the original data/parameters used to perform the mapping.
The display is clearly showing that these regions are not aligned bases, which means the BAM file is correctly annotated.
From there, I’m not sure how to help more with IGV. Maybe reset all the visualization details back to the default, load the data fresh, then start to build up the filtering again? I’m guessing that there is some other setting that is forcing those ends to be displayed. This seems to be the same advice the IGV team also gave.
I know this isn’t very helpful but the IGV team are the only people who can correct buggy display behavior. And if anyone else recognizes what is going wrong, please feel free to comment more!