I am looking for places of protein binding. To do that i need the combined.meme file from tomtom software, however I obtain empty file even if it runs succesfully. Other files return files with results



Hello @Orange_Pomeranian
Thanks for sharing the details! It sounds like you are simply not getting any hits from your query based on the current scientific parameters choices. A result of “no hits” is a valid result, so it won’t fail the tool (only actual errors e.g. seriously problematic inputs or some server issue will).
Some ideas of what to do to check the technical aspects, then to explore the scientific choices more.
-
Do you see any messages from the tool in the job logs? Find these using the pencil icon → Details tab. The log boxes will expand. This is always the first place to check whenever you get an error – but also when you get unexpected results. These are the same messages you would get if the tool was run outside of Galaxy.
-
Double check the format of the input query. You have set the options for DNA (nucleotide). Make sure the format is very simple. We can help with getting the technical details like this correct if you want to share your history back. See → How to get faster help with your question. You can unshare once we are done.
-
Try running with the defaults. Then try running with a higher evalue (less significant). What happens? This is a way to see if “no hits” might be valid for this sample.
-
Adjust other parameters to investigate. We have examples in our tutorials (also linked at the bottom of the tool form) and those link to the original author’s guide/publication, too.
-
Subset your query into smaller chunks when running these parameter tests to help them to complete quicker. See the tool Split file to a dataset collection to do this. The elements will be under the Hidden tab in your history, and you can unhide one (or, a few) to use them as inputs.
-
Finally, if you are not working at a UseGalaxy server, you could move your history over and see if that helps. See → FAQ: Transfer entire histories from one Galaxy server to another. UseGalaxy.org, UseGalaxy.eu. and UseGalaxy.org.au are the larger resourced choices.
Let’s start there! Hope this helps and if you do want to share the job logs or history back, we might be able to help with some of the above. ![]()

Great! Sharing the invocation is perfect. I’m reviewing, more soon! ![]()
Ok, I have some feedback.
The technical portions seem fine: consistent reference data throughout, valid file formats. The permission thresholds also helped the data to “produce results” but these are clearly not scientifically optimized and not significant yet.
What I would suggest from here:
-
Review the two tutorials here. Both include a workflow you could explore with your data for the latter steps, even if the data methods were different upstream, maybe the latter steps can help? Galaxy Training!
-
In particular, learn some of the characteristics of your insert sizes with this tool. I ran this on the intial BAMs in your history and see a distinct “two peak” pattern. You will want to make sure that was expected! → Hands-on: CUT&RUN data analysis / CUT&RUN data analysis / Epigenetics
-
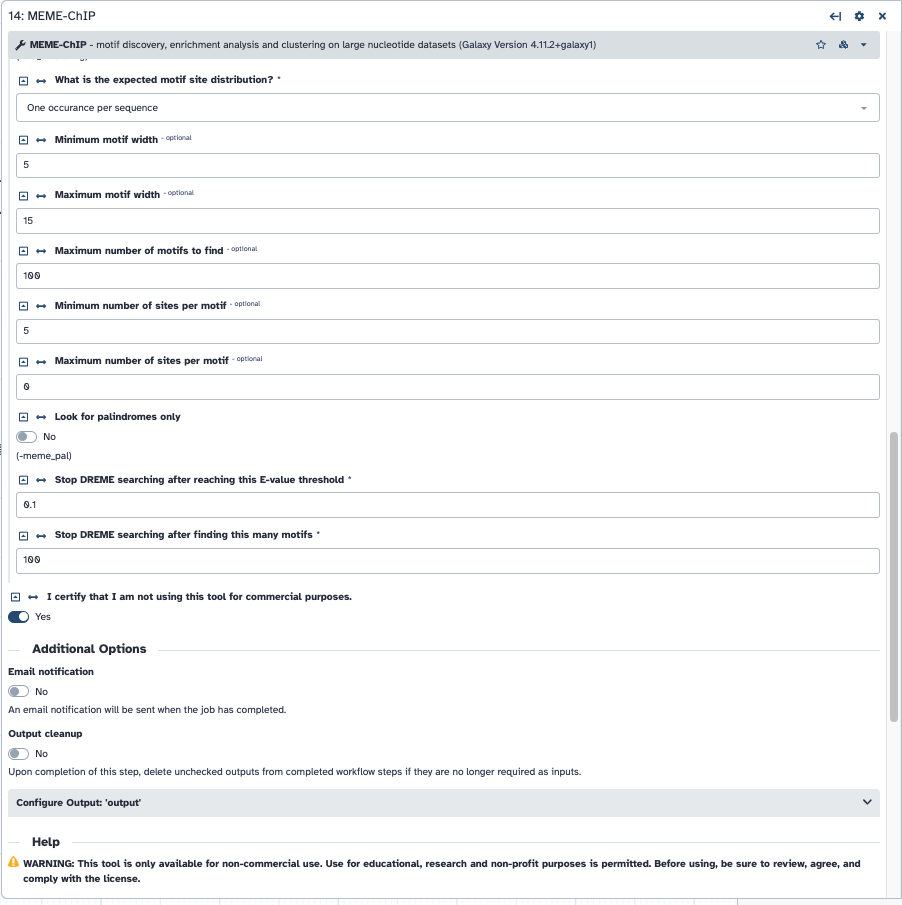
For the MEME-Chip tool specifically, your peaks are exceeding the maximum length that can be processed. This is due to changing away from the default “100” with this option: maximum size of a sequence before it is cut down to a centered section. To get an better idea of your peak sizes, you could examine the peak size (column 5 from Macs2 results) then graph as another histogram. Tools like Group and one of the Activity bar → Visualization methods could be used.
- To learn where to see this kind of message from the tool, go into your new jobs or the original job and review the warnings. This is a screenshot of your first screenshot showing where to see them. The new job’s warnings are about the lengths for some samples. The prior jobs may have something more – and given the “no hits” result, maybe everything was skipped! In short, passing “all” through to the tool “as is”" could be forcing the tool to skip them entirely, if the random sampling happened to only pick the longer peak lengths to start with. You’ll need to stay lower than that maximum length (set by the underlying tool – not just a “Galaxy” setting).
- Clicking here
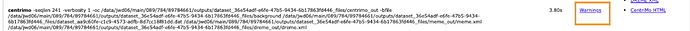
- Through to the report here
-
And, finally, consider trying to locate a publication that does something similar to what you want to do. You can also visit scientific forums to reach the people who are working with the kind of data that you are working with. We can help with technical issues here to help bridge advice between what people are doing when working with these methods outside of Galaxy into working with Galaxy, but to reach the scientists who can best help with parameter tuning, a scientific forum will reach many more people (and many will have advice already there).
- As an example, this is one discussion about the lengths and the way to tool samples. It was just the first hit when I was looking for public discussion about the longer peak lengths to reference to share! So – please be sure to review deeper than me, maybe layering in your sequencing protocol type to refine the discussion topics. → Using MEME-chip for motif discovery in sequences not derived from Chip-seq?

Hope this helps! I think you are very close to getting this working! ![]()