I’ve installed rMATS-turbo on a local server using Docker Galaxy. However, when I try to upload BAM files for sample 1 and sample 2, all files appear in both selection boxes. This seems odd because it’s unclear how Galaxy distinguishes between the two groups. I’ve tried selecting the correct samples using “Ctrl + click,” but the result still looks off.
I also tested the setup using the example data provided by the rMATS developers (Xing Lab on GitHub), but the output showed no significant splicing events — all FDR and p-values were “NA.”
Has anyone successfully run the Galaxy-based version of rMATS-turbo before?
Let’s start over in a new topic and reference the other one.
I haven’t seen the tool form as hosted in the Docker Galaxy (or, maybe forgot by now!) – but we might be able to figure this out anyway! Screenshots are probably where to start.
Try to capture these views.
The tool form. Expand the “accepted formats” toggle to reveal the datatypes each section is expecting. This can be just the “mixed up inputs” sections or more. In particular, if there are different modes that change the form, maybe capture those to reveal the different ways the tool can be used.
Datasets in the history panel. If some of the data are in collection folders, click into those and grab another screenshot, with at least one of the datasets expanded to show the metadata.
Both together. The tool form where the “potential” dataset inputs are being selected. Please make sure that the history panel is included and those same datasets are in the view (expanded, to reveal the datatype and other metadata).
It is Ok to capture a few screenshots of each with the different menus/views expanded and not, and the datasets expanded and not. You can post everything back here.
Finally, if you could confirm the tool repository and the Galaxy Docker that would help (but as I already stated, I think we’ll be able to figure it out anyway!). This one?
rMATS tool wrapper
Docker Galaxy source
I’ll watch for your reply and if you already solved this, please let us know!
Please see the screenshots below that I captured from my local Galaxy instance running the rMATS-turbo tool.
The first screenshot shows the rMATS-turbo tool appearing in the tool list after installation as Admin.
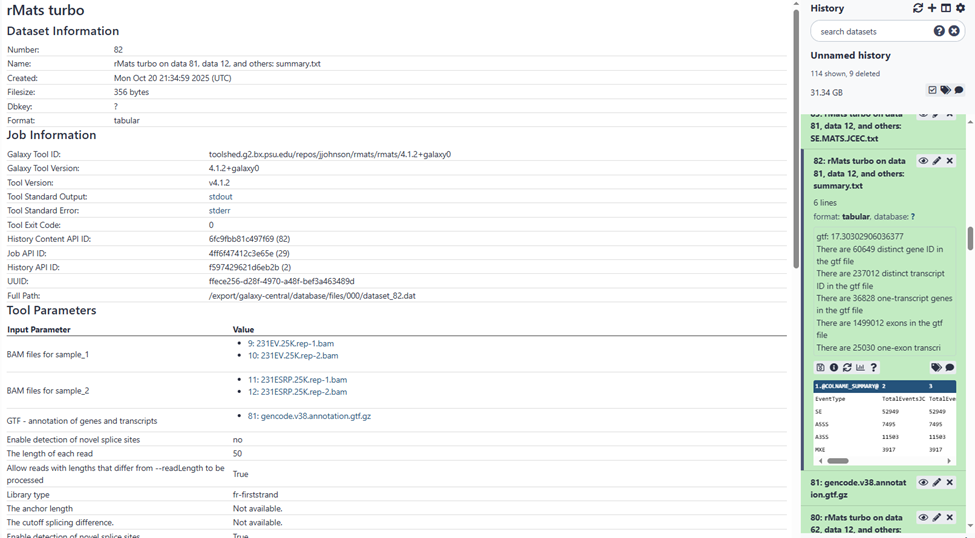
The second screenshot shows the upload interface for BAM files from sample 1 and sample 2. As I mentioned, all uploaded BAM files appeared in both boxes. I tried selecting the correct files for each sample using “Ctrl+”, and it seems to have worked based on the third screenshot, which shows the analysis details—indicating that the correct BAM files were used for sample 1 and sample 2.
The fourth screenshot shows part of the dataset list in the history panel.
The final screenshot shows my Galaxy Docker information.
On the tool form, the select field will list all potential input datasets. These will always be datasets present in the active history and with an assigned datatype that matches one of the “accepted datatypes” for that input field.
Then, from the listing, you can select the input datasets to actually use by clicking on them from the listing pull-down menu.
And to review the inputs and parameters that were applied for a job, reviewing the job’s Details view will have a table of those settings. You have this in one of your screenshots. This is a screenshot of your screenshot of the relevant section:
You are also using a gtf dataset that was sourced from Gencode that is based on the Homo Sapiens GRCh38 hg38 assembly. This is your reference annotation.
It seems you have selected two replicates from two different samples for a total of four bam dataset files.
We can’t see which reference genome you used for the mapping step to create the bams. I’m guessing this was a custom genome fasta file from the history? This should be fine.
If it was the assembly from Gencode based on the same release as the annotation, or the hg38 assembly from UCSC, then all of your reference data will be using the same coordinate system along the basepairs of the chromosomes. This is important, since this tool suite is comparing coordinates in the bam data against the gtf data to generate the results.
If the reference genome assembly was instead sourced from NCBI or Ensembl, then this is one place to double check for potential input problems that can lead to unexpected results. The guide here explains with more details. → Reference genomes at public Galaxy servers: GRCh38/hg38 example
Other than that, different parameters can impact the results of course! The original manual from the tools and potentially online discussion at a scientific forum like Biostars.org are the resources to review to learn about fine tuning for specific outcomes. The tool will work in Galaxy about the same as anywhere else. Or, should. If you notice some bug with the tool wrapper, you can report this to the developer at their Github repository.
Thank you, Jennaj! I downloaded the bam files directly from GitHub -Xinglab/rmats-turbo. I will check again whether I am using the correct reference gtf files for analyzing their test data.