I’m testing just one PE sample, not tagged, and I have no idea why MACS2 callpeak is showing error.
History: Galaxy
Thanks so much!
I’m testing just one PE sample, not tagged, and I have no idea why MACS2 callpeak is showing error.
History: Galaxy
Thanks so much!
Hello @candyc
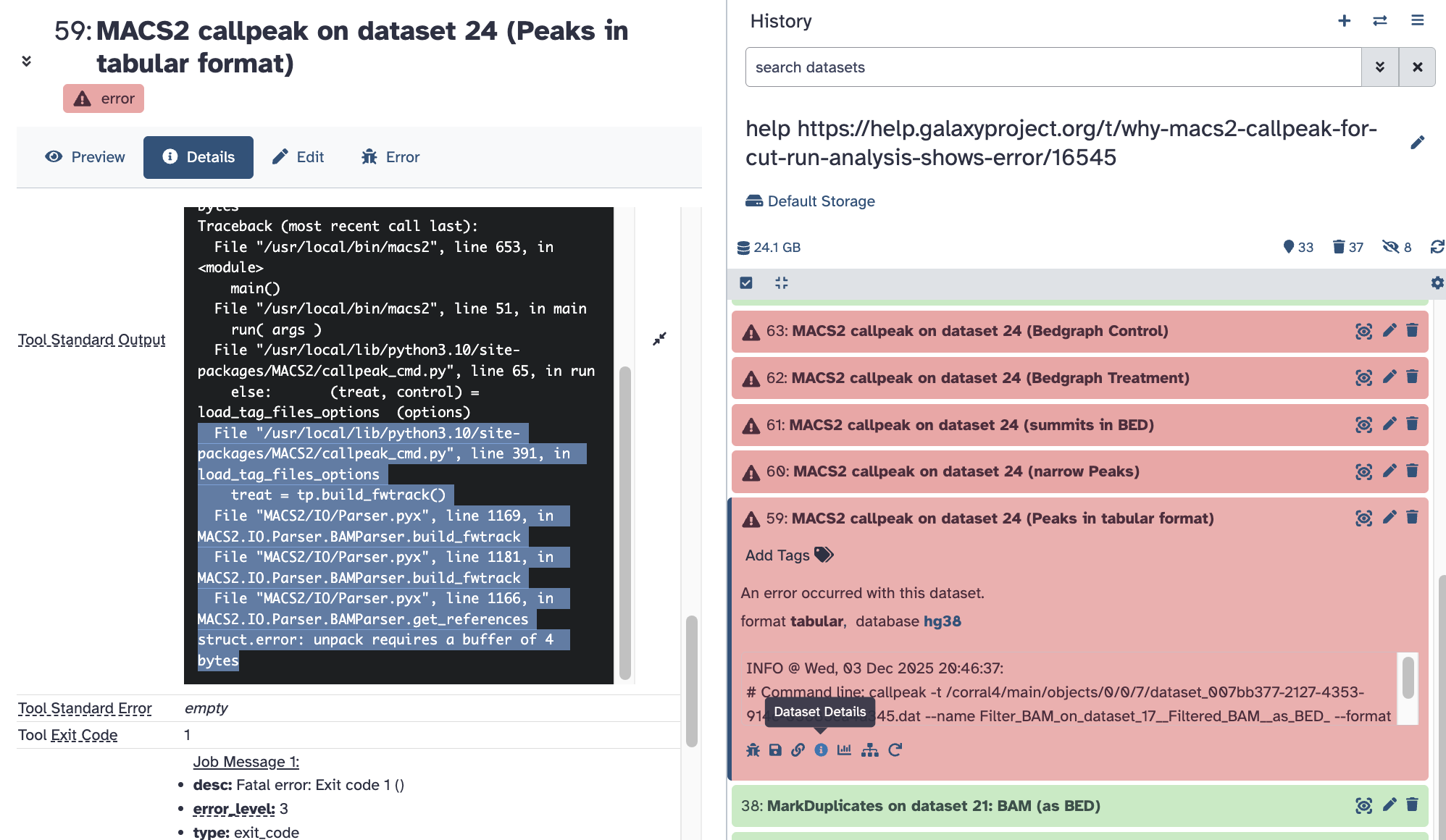
Thanks for sharing the history!
Yes, this error is a bit confusing. The tool is complaining about unexpected data content within the genome index. This comes from MACS2 callpeak itself and indicates that the tool found unexpected mapping positions it couldn’t parse when building up the internal data structures.
This can sometimes be from a genome mismatch but for your case, it has to do with including the haplotype/fragments in the genome when mapping. Using the canonical reference instead is the solution.
![]() GTN Example → Hands-on: CUT&RUN data analysis / CUT&RUN data analysis / Epigenetics (#hands-on-mapping-reads-to-reference-genome)
GTN Example → Hands-on: CUT&RUN data analysis / CUT&RUN data analysis / Epigenetics (#hands-on-mapping-reads-to-reference-genome)
You may get a slightly different result between the two, since the original competition for a mapping position will be a bit different. But that should be minor due to the other strict mapping quality filters applied. Your choice!
Remap the reads against the hg38 canonical reference genome.
Or, you can use one of the filtering tools on the BAM or BED files to restrict the reported regions to the primary autosomes + sex chromosomes (and sometimes just chrX).
Please give that a try and let us know if it helps! ![]()
Hi @candyc
Great! Thanks for giving that a try.
It seems the tool is still getting overwhelmed by the volume of reference data, and while I think your results are technically correct so far (those hits are valid but numerous), filtering the data down a little bit more to get rid of the fragments might help! Cut&Run produces a more complicated analysis for MACS2 so .. doing whatever you can to give the tool only what will end up processed tends to work better (reduces the load on the tool).
I’ve run the other steps you were originally applying on your new data and started up MACS2 again. Let’s see what happens!
If I somehow missed those filtering steps applied to your original rerun, my apologies. I think a rerun will still have some utility, so, let’s see what happens! ![]()