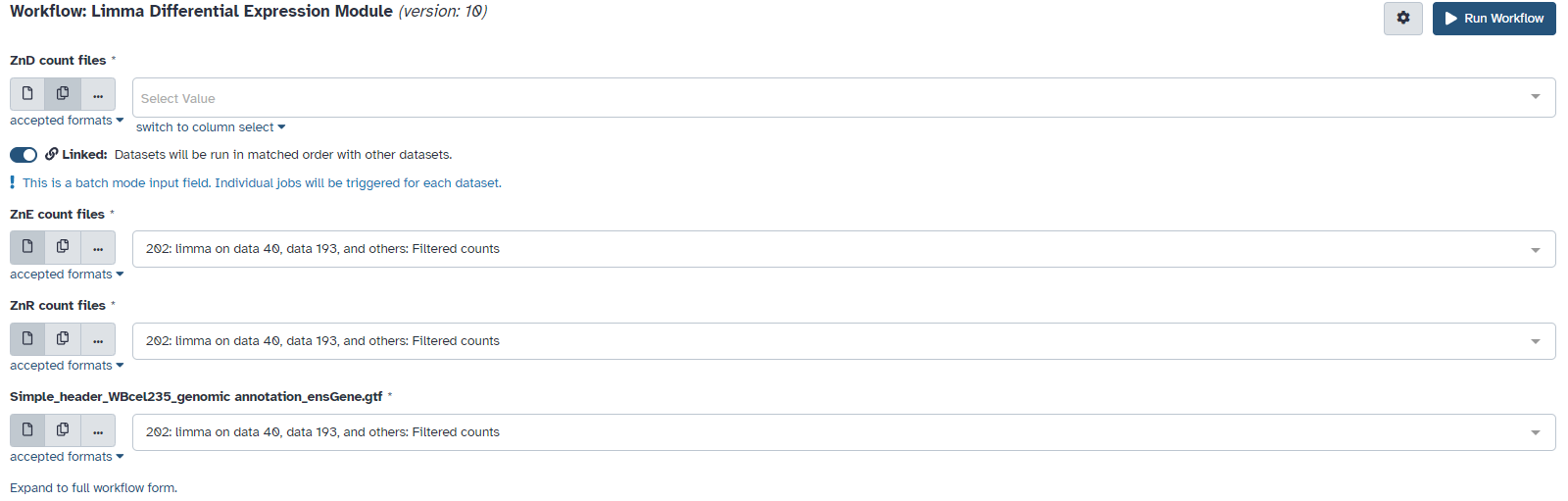
I have a workflow set up for limma. In the original limma module, I have three biological replicates for each group, and I was able to select the three files from my history using first “multiple datasets” button and then the “…” button. After I saved this as a workflow and run it, file input stops working. I was able to select from the list by clicking “multiple datasets” and then “…” next to it, but the selected files don’t go into the menu unless I put the files in a collection first
Is this intentional? It’s a bit funny since there’s also an “input data collection node”.
Also, I’m not sure I understand the “linked” checkbox under each input dialog. My understanding is that limma uses the three files in the input as a group, so that for files a, b, c in group 1 and d, e, f in group 2, it’ll run (abc) vs (def). The “linked” checkbox makes it sound like the workflow will run limma three times, with a vs d, b vs e, and c vs f each time, and “unlinked” will be running it 9 times with a vs d, e, f and b vs d, e, f and so on.
Hi @billy.l
It seems you submitted the workflow in batch mode. As the description says, This is a batch mode input field. Individual jobs will be triggered for each dataset.
This is for linked data. For unlinked data you get “all against all” submission.
Workflow with collection might be the best option. Without collections you have at least two options. A workflow with fixed number of inputs, eg 3 vs 3 (example) or do not use input box (example2).
Hope that helps,
Kind regards,
Igor
Thank @igor. You’ve been really helpful in the past days. I really appreciate it!
If I chose input as collections, the genes don’t get proper annotations in the results; if I use fixed number of inputs, the tabular files still don’t get registered if I choose them; not using input box won’t allow me to choose multiple files. Hiding and unhiding the hidden tabular files don’t help, and since the files are directly from featureCount and then replace text (to fix the gene version generated by ensembl, eg. “XXXX.1”), I’m sure they are tabular. I think there is a bug with input in limma in the workflow mode
Rerunning the same limma module by clicking the refresh icon from history and then emptying the input selection boxes also gives me the same problem. I can select the files using the “…” button but they don’t show up. Dragging the files into the box doesn’t work, either.
I am a regular user on ORG server, not admin, no special privileges.
What browser do you use? Try Firefox.
Click at pull-down menu to select the input datasets.
Yeah. The problem is that when I use your methods with my workflow and history it doesn’t work. The workaround is start the module anew from tools, or rerun limma from a different history then switch to my current working one.
Hi @billy.l
The workflow looks OK, but I am concerned with (apparently) a gtf file used as gene annotation (based on name of the corresponding input box). I believe limma uses output of AnnotateMyID. See Hands-on: 2: RNA-seq counts to genes / 2: RNA-seq counts to genes / Transcriptomics
Can it be the issue? Does it work if you remove the annotation input?
Also, description for factor name says “Please only use letters, numbers or underscores.” while you used a dash “-”. Try CamelStyle, ZnContent.
Hope that helps.
Sorry, that was a label problem. The gtf file is for mapping and featureCounts. The gene annotation file is actually “Ce11 RNAseq annotation BioMart.tsv”. When I rerun the workflow or from history, the correct annotation file is recognized automatically.
Removing the dash in factor name didn’t help. It’s funny, because originally I have a space for the dash. Thanks for your help though! I’ll find time to submit a bug today.