Hello,
I am currently trying to analyse some methylation sequencing data using Bismark. When I try to extract the methylation status using the Bismark Meth. Extractor, I am expecting to also get a .cov coverage file. However, this does not appear to be being generated.
The tool output gives the following:
Writing bedGraph to file: dataset_e734b08e-b355-49c5-94cc-61ef7b831d1e.datbedGraph.gz.
Also writing out a coverage file including counts methylated and unmethylated residues to file: dataset_e734b08e-b355-49c5-94cc-61ef7b831d1e.datbismark.cov.gz
However, I do not see the cov.gz in the resulting result archive. Am I using an incorrect setting (I had asked it to give me the genome wide cytosine report) or is this something that it currently does not output?
Any help would be greatly appreicated!
Many thanks,
Lewis
Methylation proportion report for each possible position in the read (Mbias Report)
This report shows the methylation proportion across each possible position in the read (described in further detail in:Hansen et al., Genome Biology, 2012, 13:R83). The data for the M-bias plot is also written into a text file and is in the following format:
read position | count methylated | count unmethylated | % methylation | total coverage
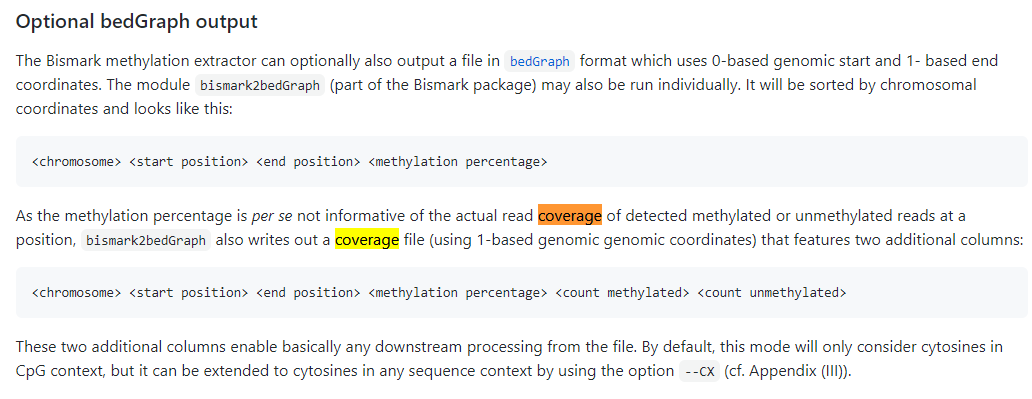
Many thanks for your reply! The M-bias report is not quite what I am after. This report is to check whether there is any bias in methylation across the read. As I did 150 PE sequencing, this outputs a file for each one of the 150 positions. What I am after is a coverage file that has counts (both methylated and unmethylated) and their genomic position. The methylation extractor should give this as a .cov file, but this doesn’t appear to be being saved. Here is a screenshot from the Bismark manual of what I am after:
The *cov.gz file declared at the output log is only created as a temp file in order to build other outputs;
Writing bedGraph to file: dataset_f5b173bc-5683-4e3a-aae9-bbfab0223a5b.datbedGraph.gz
Also writing out a coverage file including counts methylated and unmethylated residues to file: dataset_f5b173bc-5683-4e3a-aae9-bbfab0223a5b.datbismark.cov.gz
(…)
Output will be written into the directory: /data/dnb03/galaxy_db/job_working_directory/022/607/22607745/tmp/tmp7xnxvb6j/
Summary of parameters for genome-wide cytosine report:
Coverage infile: dataset_f5b173bc-5683-4e3a-aae9-bbfab0223a5b.datbismark.cov.gz
Output directory: >/data/dnb03/galaxy_db/job_working_directory/022/607/22607745/tmp/tmp7xnxvb6j/<
Parent directory: >/data/dnb03/galaxy_db/job_working_directory/022/607/22607745/tmp/tmp7xnxvb6j/<
Genome directory: >/data/dnb03/galaxy_db/job_working_directory/022/607/22607745/tmp/tmpeuc9ajoz/<
CX context: yes
Genome coordinates used: 1-based (default)
GZIP compression: no
Split by chromosome: no
.
Alternatively, the output of the methylation extractor can be transformed into a bedGraph and coverage file using the option --bedGraph (see also --counts ).;
which I can’t see in the command line of my tests:
Previsously, this error was solved and related to another optional parameter, buffer size (--buffer), in standalone bismark_methylation_extractor v0.20.0; But I can’t test this now.
I totally agree with you that it seems the .cov file is not being saved, and instead is being used only as a temporary file to generate the genome-wide cytosine coverage report. The --bedGraph option is definitely there in the command line code you copied above, so I don’t think that this is the issue. The bedgraph file (.datbedGraph.gz) is present in the results archive generated when asking for the genome-wide cytosine report, which is why I found it odd that the .cov file is not there as well. It would definitely be interesting to see if the --buffer option is the cause of this file not being present, however if it can’t be fixed then I think I’ll most likely have to find another way of running the bismark extractor! Many thanks for your help so far!
Ah I see, that definitely could be it! Is there any way of testing this theory out? I have no idea who to contact to see if the methylation extractor could be changed or modified on Galaxy to be able to produce the .cov file, or even if this is possible!
@David and @gallardoalba many thanks for all of your help and input, and for updating the extractor to provide the coverage file. I’ll make sure to look into Methyldakel if you recommend this as a better way of getting the methylation statistics.