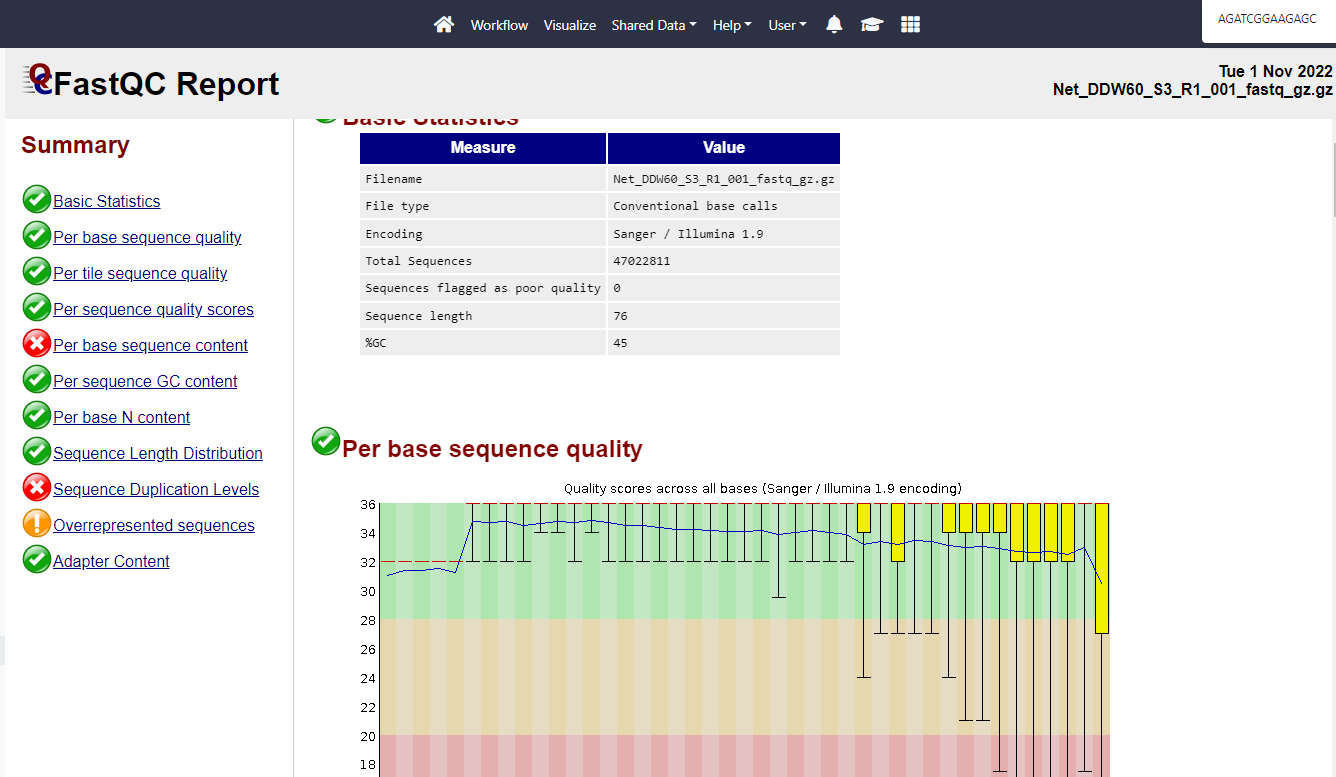
Hello.
I’m attaching a snapshot of the fastQC output of a sample, I’m debating how to QC this and similar samples, on the one hand it look alright as is, on the other hand I sent it to bioinformatic and he did the following:
Trimming and filtering of raw reads
Raw reads (fastq files) were inspected for quality issues with FastQC. Following that, the reads were quality-trimmed with cutadapt, using a quality threshold of 32 for both ends, poly-G sequences (NextSeq’s no signal) and adapter sequences were removed from the 3’ end, and poly-T stretches were removed from the 5’ end (being the reverse-complement of poly-A tails). The cutadapt parameters included using a minimal overlap of 1, allowing for read wildcards, and filtering out reads that became shorter than 15 nt. Finally low quality reads were filtered out using fastq_quality_filter, with a quality threshold of 20 at 90 percent or more of the read’s positions.
his QC caused the lost of 16% of the reads
I also triad using fastp on default setting which only discard 1.2% of the reads…
i will be happy to hear your inputs
Thank you
Netanel