The results of HTSeq is shown in plain image . What could be reason for this ?? Erroneous Gff file or erroneous Bam file? I am attaching here sorted report of the bam file ( green background) as well
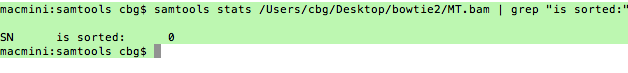

The results of HTSeq is shown in plain image . What could be reason for this ?? Erroneous Gff file or erroneous Bam file? I am attaching here sorted report of the bam file ( green background) as well
Does the GTF dataset contain any data lines with “exon” in the 3rd column? The error suggest that it doesn’t and you either need to modify the feature used for counting or find a more suitable GTF source.
Exon is the default feature considered by HTseq-count wrapped for Galaxy. Screenshot:
Please see the RNA-seq tutorials for example usage:
Support FAQs:
