Recently, I submitted a job to annotate a genome using the BRAKER3 tool. The job ended with an error described as follows. Could you please check into this? I need to get this annotation as soon as possible. I believe this is a permission issue that the Galaxy/BRAKER3 admin needs to check.
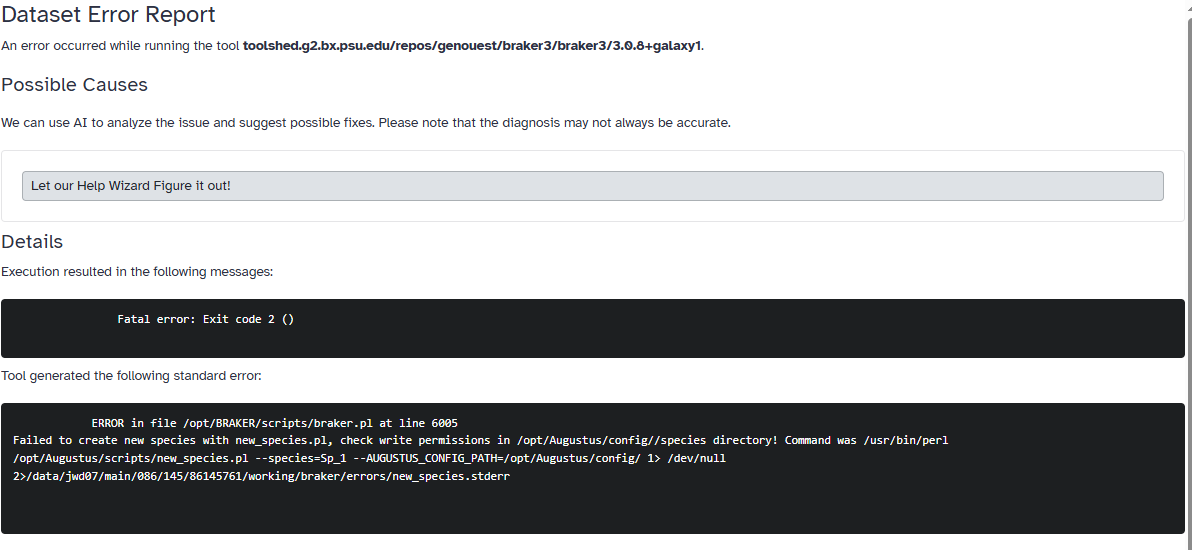
Fatal error: Exit code 2 ()
ERROR in file /opt/BRAKER/scripts/braker.pl at line 6005
Failed to create new species with new_species.pl, check write permissions in /opt/Augustus/config//species directory! Command was /usr/bin/perl /opt/Augustus/scripts/new_species.pl --species=Sp_1 --AUGUSTUS_CONFIG_PATH=/opt/Augustus/config/ 1> /dev/null 2>/data/jwd07/main/086/145/86145761/working/braker/errors/new_species.stderr
Would you be able to confirm that you are working at UseGalaxy.eu? And, are you following a tutorial (this one?) or is this your own data.
I’m also starting up an independent test at the EU server with the tutorial data to see if I can trigger the same error, but if you would like to capture the Job Details table for your run (lists all runtime parameters) that would be very helpful! A peek at your genome file would be helpful too (also on that view, the expanded dataset is enough since it will show all the metadata).
Use the i-info icon on any dataset, or tab into Details from the bug report view.
Let’s start there – I’ll get my simple test going meanwhile. Thanks!
XRef
Not all of the available parameters for BRAKER3 can be supported at the public Galaxy servers. Details → genome annotation with Braker3
# Tue Jul 15 19:37:34 2025: Log information is stored in file /data/jwd07/main/086/145/86145761/working/braker/braker.log
Tool Standard Error
ERROR in file /opt/BRAKER/scripts/braker.pl at line 6005 Failed to create new species with new_species.pl, check write permissions in /opt/Augustus/config//species directory! Command was /usr/bin/perl /opt/Augustus/scripts/new_species.pl --species=Sp_1 --AUGUSTUS_CONFIG_PATH=/opt/Augustus/config/ 1> /dev/null 2>/data/jwd07/main/086/145/86145761/working/braker/errors/new_species.stderr
Tool Exit Code
2
Job Messages
* desc: Fatal error: Exit code 2 ()
error_level: 3
type: exit_code
exit_code: 2|
|Job API ID|11ac94870d0bb33a8a436ffdfc0a24f1
Thanks for sharing the parameters, and I think I see the problem.
If you can supply the input for
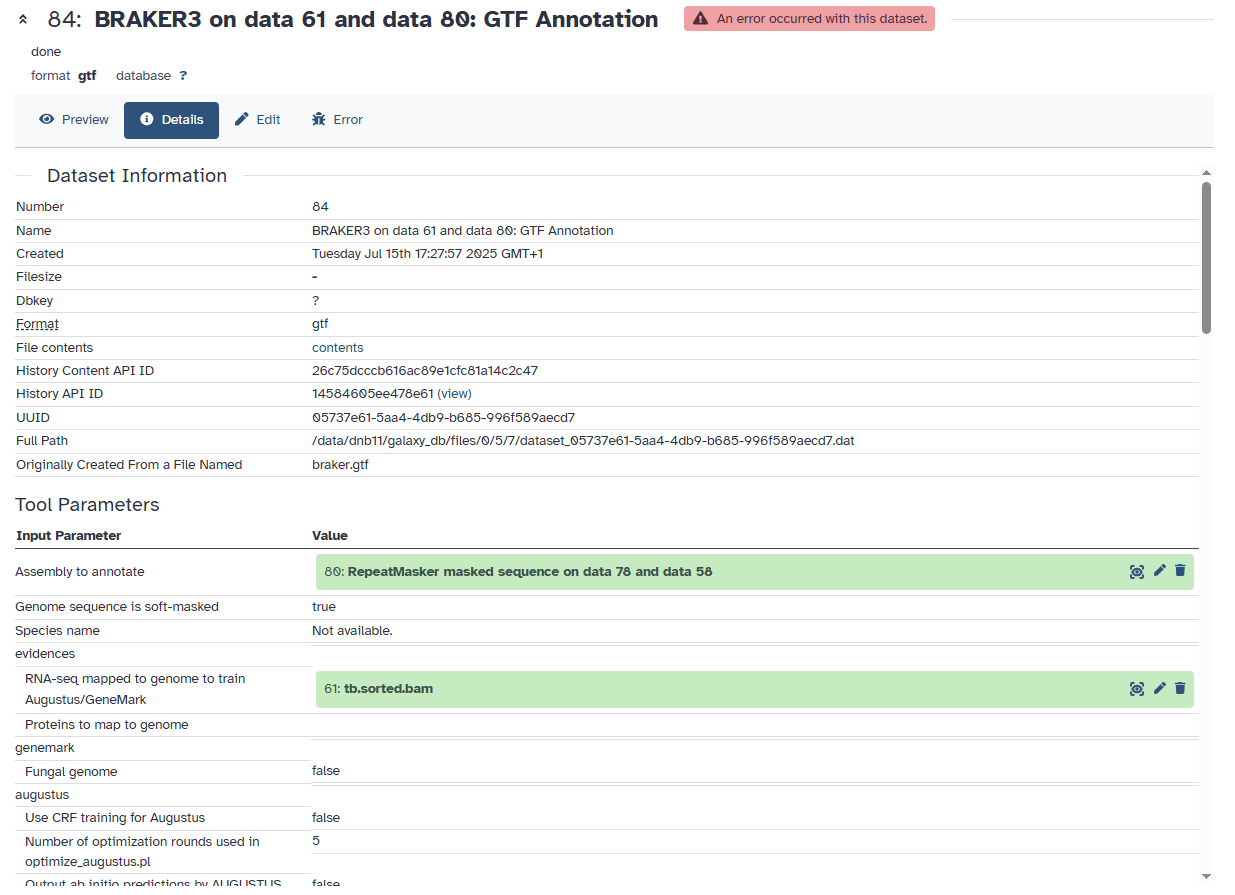
RNA-seq mapped to genome to train Augustus/GeneMark
Then, there will be data available for this option to also be toggled to true
Output alternative transcripts based on explicit evidence from hints
If not supplied, then the tool is falling back and attempting to call using GeneMark directly, which isn’t available in the public version. The CFC predictions are also not available from Augustus, but with the BAM mapping you can use ab initio predictions.
A bit complicated but hopefully this helps! I don’t think this has changed from what was available in the prior versions hosted at the EU server (and the GTN VGP tutorials also support this usage), but if you can share an example, we can look into it more with that exact comparison. I didn’t have any successful jobs with the other options used.
Hi @jennaj,
I had supplied the RNA-Seq mapped bam file for this job to train Augustus and GeneMark. However, I was still having this error. In the past, I had obtained annotations without providing any BAM file, too.
I just noticed when you mentioned aboout the RNA-Seq file.
Somehow when I copied and pasted the job details, the file names were not copied which I was using for the job. Below is the screenshot which shows my file names that were used for this job which ended up having an error. You can see that I have used RNA-Seq mapped bam file.
I am not expert in this, but the error suggests that there is a write permission issue when writing it for new species. As i need a annotation for a species which is not in a database currently, I can not chose its name from the drop down menu.
I am not sure, but may be a recent update of BRAKER3 did not have write permission to public. Granting a write permission to all would might solve the issue?
Please let me know when this will be fixed. And thank you both of you for checking this issue.