Welcome @erdavenport
Thanks for sharing all these details, and I think we’ll be able to help!
There are two similar tools for NCBI SRA fastq data download. The underlying scripts are based on the same NCBI’s toolkit but the options are configured to work slightly differently.
I’m going to define the tools first, then address your question. Feel free to jump to the bottom then use the extra help as a reference instead. 
Tool Help
What it does?
This tool extracts data (in fastq format) from the Short Read Archive (SRA) at the National Center for Biotechnology Information (NCBI). It is based on the fasterq-dump utility of the SRA Toolkit. The following applies:
- if data is paired-ended (or mate-pair) the tool will generate a collection of file pairs, in which each element will be a pair of fastq files containing forward and reverse mates.
- if data is single ended, each element of the collection will be a single fastq dataset.
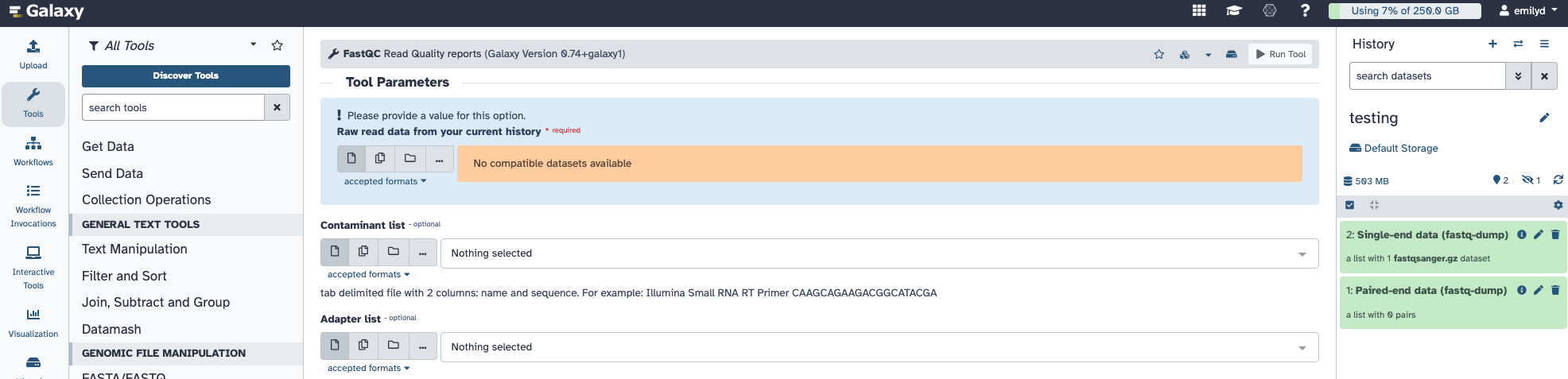
Both tools have the same initial Help section but the data is organized a bit differently between the two types of output! The collection folder shape is written on the collection folder in your history. updated
These are the main differences:
Download and Extract Reads in FASTQ format from NCBI SRA (current versions) and Faster Download and Extract Reads in FASTQ format from NCBI SRA
Collection folder shape: list of pairs
List of pairs collections are a nested listing of files.
-
Each sample will be written into a grouped element representing that sample. The dataset files will be inside of it.
-
If the data is single end, each sample will have one fastq file.
- One file of R1 reads per sample.
- The sample group is at the first level, and the fastq file is nested inside of it.
- Later on, if the data is sent to a tool that produces multiple outputs, each will still retain that top level sample element identifier (and any group or name tags added).
-
If the data is paired end, each sample will have two fastq files.
- One fastq file for the forward (R1) reads and one for the reverse (R2) reads.
- The sample group is at the first level, and both fastq files are nested inside of it.
- This structure is understood by tools that can accept a paired end collection shape and can considerably speed up analysis.
- The sample element identifier (plus tags) stay with that pair throughout the analysis.
- With one sample pair or hundreds (!), and maybe complicated sample/condition tagging, this organization can be very powerful.
- Blog from last week with a video explaining how this works! → Why collections?
 Download and Extract Reads in FASTQ format from NCBI SRA (legacy versions)
Download and Extract Reads in FASTQ format from NCBI SRA (legacy versions)
Collection folder shape: list
 * This is an update! Current versions of both tools will work the same at the UseGalaxy servers. Other servers may still host the legacy version.*
* This is an update! Current versions of both tools will work the same at the UseGalaxy servers. Other servers may still host the legacy version.*
List collections are a flat listing of files.
- Each sample will be written into a single element of a list collection.
- If the data is single end, each sample will have one fastq file of the single reads.
- One file of R1 reads per sample.
- If the data is paired end, each sample will be a fastq file containing both reads together.
- One file with both the R1 and R2 reads per sample.
- This is the “interleaved” type of fastq file – many tools will expect that these will be split out into the forward and reverse reads. The tools from the Seqtk tool group are one way to do this.
So, that is a lot of information! But maybe helps to explain why data in “a collection” might not be enough when choosing how to send data to tools. The datatype matters, but also the shape of the data. You are never stuck in any particular shape! See Collection Operation tools for more.
I see you are using the first. The collection folder shape list is accepted by FastQC. You should be able to connect this to tool. However, because the downstream tools are connected first and this can lead to some workflow metadata conflicts. If you hover over the connection, you will get a message about the “shape” of the collection not being a match for what is expected.
 If there is more than one sample in your list, remember to include Collection Operations → Flatten Collection to combine the sample name with the read end label into a unique label (element identifiers) → sample_forward (single end) or sample_forward and sample_reverse (paired end). This will give each fastq dataset’s report a unique label when these are later combined with a tool like MultiQC.
If there is more than one sample in your list, remember to include Collection Operations → Flatten Collection to combine the sample name with the read end label into a unique label (element identifiers) → sample_forward (single end) or sample_forward and sample_reverse (paired end). This will give each fastq dataset’s report a unique label when these are later combined with a tool like MultiQC.
Try this:
- disconnect all the workflow connection noodles
- then, connect the tools again in the order of execution
- for your use case, you will be connecting just one of the outputs from the Download and Extract Reads – the single end list connection
- once connected, this informs each downstream tool that a list collection of data is passing through
- you might need to adjust some of the input parameters on the other tools, too
- this is an excellent tip to remind students about! If the noodle don’t connect, reset the workflow metadata by disconnecting all, then connecting from the start. I usually say that getting the inputs on the canvas as the first step is a good idea!
Please give that a try and let us know if it helps or not! You are also welcome to share back your workflow, or a failed workflow invocation, and we can troubleshoot this more if simply reconnecting is not enough. Maybe one of the downstream tools need an input choice adjusted. Thanks! 
BONUS 1
For an example of a similar QA workflow that can handle a paired collection without running into duplicated sample identifiers with FastQC/MultiQC, please see this post. The example workflow could be adjusted to work with single end reads too! This one also has some simple subworkflows that might be good for followup for intermediate students.
Then, this is one discussion from last week where we went into more detail about why this organization works! → Issues with receiving results from FastQC in MultiQC from several collections of samples - #5 by jennaj
BONUS 2
One tip I might suggest is to add in another input! This could isolate the accession input to a single field on the workflow runtime form (at the top!), or a separate text file with a list of accessions (selected from the history). This could be annotated with a usage warning “Single end samples only!” but stated a bit nicer! 
.