Dear @jennaj,
Thank you very much for your advice. I have now tried running both Salmon and DeSeq2 a few more times, but the issue remains.
Double check that these settings are correct:
- Choice of Input data → TPM values (e.g. from kallisto, sailfish or salmon
- Program used to generate TPMs → Salmon
- Gene mapping format → input the same annotation (GTF or tabular) as input to Salmon.
If you don’t have item 3, you’ll need to back up and rerun Salmon with that annotation to produce the proper inputs for DESeq2.
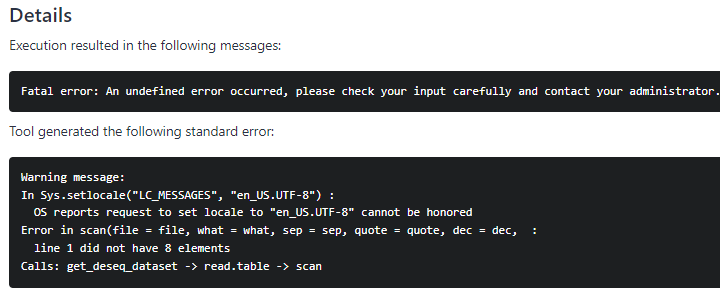
Yes, the first run was wrong. I now corrected 1 and 2. Unfortunately, for 3. I only have tabular data from BioMart. I reran everything several times, but issue remained. I removed the title row from the transcript/gene ID table, the error message changed to the following:
Fatal error: An undefined error occurred, please check your input carefully and contact your administrator.
Warning message:
In Sys.setlocale(“LC_MESSAGES”, “en_US.UTF-8”) :
OS reports request to set locale to “en_US.UTF-8” cannot be honored
reading in files with read.delim (install ‘readr’ package for speed up)
1 2 3 4
Error in $<-.data.frame(*tmp*, “TXNAME”, value = character(0)) :
replacement has 0 rows, data has 274080
Calls: get_deseq_dataset → $<- → $<-.data.frame
Please be aware that DESeq2 expects all sample counts to be in individual dataset inputs. This error might come up if a matrix of counts was input. The “8 elements” is not expected – that number of columns does not match the column count of any of the expected inputs:
- quant.genes.sf files has 5 columns and “a single header or not” is specified on the tool form
- transcript to gene has 2 columns and should not contain any header lines (# or others)
- GTF has 9 columns and should not contain any header lines (# lines).
I think my input was individual datasets (nothing was merged, concatenated, etc.). Also, I don’t have any GTF data, but the formats of other datasets seem to be correct and the number of columns seems to match.
As advised, I sent the bug report.
These are the datasets I used for the most recent round of runs (the ones that resulted in altered error message):
Salmon:
Transcripts fasta file: 9 (Fasta normalised reference transcriptome).
Input datasets: paired data, 1-8 (trimmed reads; 1,3,5,7 at Mate pair 1; 2,4,6,8 at Mate pair 2).
File containing a mapping of transcripts to genes: 10 (transcript ID-gene ID table from BioMart. I think I changed it to tab-delimited. I also took off title row).
Outputs were 11-18.
DeSeq2:
Comparison group 1: datasets 12 and 16.
Comparison group 2: datasets 14 and 18.
Gene mapping format - tabular.
Tabular file with Transcript-ID to Gene-ID mapping: dataset 10 (the BioMart table, with transcript and gene ID versions).
If full history is required, I would prefer to share it privately.