I’m following along the RNA-Seq reads to counts tutorial with my own data and am up to the Generating a QC Summary Report step.
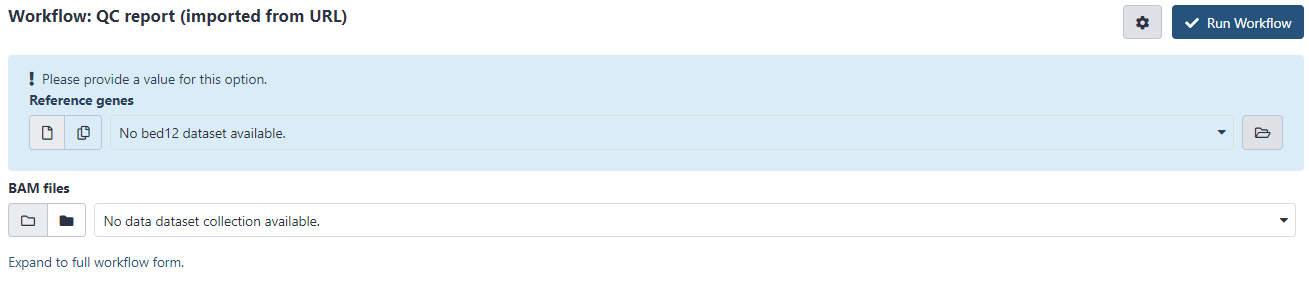
I’ve followed the instructions (imported the QC report workflow, downloaded the RefSeq BED file) but in the workflow I cannot seem to select my BAM files - it only gives me the option to select MultiQC files.
It is a bit hard to debug for me so I hope someone else can give a better answer . But if I go to the workflow page after getting the workflow I see this: