I was trying to use Orthofinder to find orthologous protein/gene groups across four different plant species. I dont have the blast results, so I was using Orthofinder from scratch with the protein fasta files. I tried with the protein and the cds fasta files but everytime the program ended in error stating the list contained zero datasets. I am unable to understand where I am going wrong. Could anybody please help me find a solution to this issue? Also, if instead of using the “from fasta files” option, I want to use the “from blast results” option, then how should I proceed with that? How to obtain the blast results before running orthofinder? Any help, any tutorial, any suggestion is please welcome. I am stuck with this problem for more than a week.
I’m curious about which server you are you using the tool, and what your files look like. Would you like to share your history so we can help you to troubleshoot the usage? Please leave the errors or odd results that you have so far in history undeleted if possible. The link can be posted back in a reply and you can note which datasets represent the different ways you have tried this along with some explanation about what the goals are.
How to share your history is in the banner at this forum, also here. → How to get faster help with your question.
Let’s start there, thanks! ![]()
This is my history. Initially, I was not sure whether I should use protein fasta files or genomic DNA fasta files to run the all against all blast program in OrthoFinder. So, I used both the files. Jobs run from 1552 to 1565 are done using the genomic DNA files of four plant species, while jobs from 1594 to 1607 are done using the protein fasta files of the same four plant species. In the second case, no gene trees were obtained (job no. 1596). I obtained results for both the cases. Now, after running this blast, I tried to input these results in OrthoFinder again using the “from blast results” option (job ids 3547 to 3561, 3562 to 3576, and 3577 to 3591) using different options. But all these jobs failed. My main aim is to obtain a Venn diagram for these four species based on the shared and unique orthogroups as seen in published manuscripts. I am not aware as to how can I obtain such diagrams and tables?
Thanks for sharing the history, this helps.
Some feedback I noticed from the first error I looked at
-
Click into the i-icon for the errors, and review the messages. Most of these are about the content of the inputs being a mismatch for what the tool is expecting.
Example: Data 1658.
The message is explaining why the tool thinks the annotation is not in the expected format with details. Then, when reviewing your inputs, I can see that some of the annotation files are not in the GFF3 format, but GTF and a hybrid type of GFF.
All must be GFF3. You can use the tool gffread sometimes to covert between these formats but if the data source has a GFF3, get it directly from them.
-
Organize the data into collections. If you are supplying genome and annotation pairs, those need to be read in by the tool in the same order. Two collections with the same order would help.
More feedback soon. I am still running a separate test on one of these tools to confirm a different problem you had.
Update! The tests completed. I ran some smaller genome files through the two tools you were using to make sure the technical parts were Ok. Everything worked. I’ve shared the history here. I didn’t use all parameter combinations, so if you get an error with this data and specific combinations, you can share that back for more feedback.
Meanwhile, you should explore the user guide for the tool linked from the Help section. This should have example data you can compare to. ![]()
Thank you for your help. I am trying to rectify the gff files before running OrthoFinder and Proteinortho again. While running Orthofinder using the protein files, I did not get any gene trees; remaining output files I received except the gene trees (job no. 1596). Is it because of issues in gff files or something else?
I saw you provided some results of Proteinortho as well which I couldn’t. Is it because of the same issue in gff files? For Proteinortho, I didn’t get a single output.
Apart from this, since you are helping so much, do you know how I can find out what are the single copy and multi copy genes from these four plant genomes? I wish to segregate the single copy, double copy, triple copy, and multi copy genes. Can you please help on this aspect as well? ![]()
Yes, the format and labeling of the input data is likely contributing to the problems.
That is what I meant with these comments:
What to do
Summary:
- You should create or retrieve the correct type of GFF3 annotation, then put those files into a collection folder.
- Simply the format of the fasta files, then put those into a collection folder.
- The order of files in those two different collection folders should be the same: data for genome A is listed first in both, genome B second, … etc.
Formats:
-
GFF3 from the data provider should be already in the correct format. If you only have GTF, you can try to convert it to GFF3 with gffread. See → Datatypes - Galaxy Community Hub
-
fasta for the exomes needs to be very simple: just have the identifier on the > title lines of the fasta, and remove everything else. This usually means removing everything after the first whitespace on the > lines, and the tool NormalizeFasta can be used. See → Datatypes - Galaxy Community Hub
-
Finally, double check that all of the identifiers in the fasta are now actually inside of the GFF3 files. If not, then you do not have the correct data for one of these and you still need to find or create the paired files.
- The tool is trying to “match identifiers up” between the files. It uses the protein sequence to do alignments, then it uses the identifier from the fasta to look up annotation details in the GFF3 to do some clustering.
- You will want to make this very clear and exact – not just to have the tool run without failing but to get accurate technical outcomes in the results. You can’t make any scientific interpretations about the annotation without this.
I noticed that your runs without the annotation seemed to work Ok, but that is because the tool was only using the protein fasta sequences against each other, not really using the fasta identifiers for anything but labels. But when bringing in more data files to consider, you will need to make sure the tool can first make associations between both files per exome, then layer in the clustering between the species’ exomes.
Exact suggested steps
- Copy all of the fasta and GFF/GTF/GFF3 files you have into a brand new history.
- Then start going through each species’ files – clean up the fasta file, then decide in the annotation is actually GFF3 or not, and fix that up.
- Once you have done this for all, create a list collection for the fasta’s, then one for the annotation.
- Finally, I would copy just those final two collections into another new history and try a rerun using this very clean data.
If you are not sure how to manipulate collections, this tutorial has examples.
- Hands-on: Using dataset collections / Using dataset collections / Using Galaxy and Managing your Data
- Collections are just a special kind of folder: these link special copies of your data into a type of data structure that makes it easier for tools to process data in batches, but also in logical groups. You are mostly interested in the logical groups for this tool.
Once you have the files cleaned up and if you need help with getting these into collections, you can share back that history and we can try to help more. ![]()
I tried to rectify the protein fasta files and the gff files. I used the option NormalizeFasta to convert the Fasta files and then I ran OrthoFinder but still I did not obtain any gene trees. I downloaded the original gff3 files as well. However, still I don’t get any output using Proteinortho. I am sharing the 4 fasta and gff files here in this history. Please help me obtain the results. Also, please mention how can I obtain clustervenn diagram for the orthogroups and how can I obtain results for single copy and multicopy genes? What tools should I use? Please help.
Great, this is looking so much better!
Each of the GFF3 files need to have a line like this one at the very top. This is what tells tools that the file is actually that content, otherwise it will read it in as a generic GFF file. You can remove the other comment lines in each or just put this.
##gff-version 3
Please adjust that in your files, then try to run the OrthoFinder tool in this same history so we can see how it is working and what the new log messages state.
You are getting much closer to solving this!
I re-uploaded the gff3 annotation files with only header as you mentioned. Then I ran ProteinOrtho which did not give any result. I also ran OrthoFinder using 3 different parameters; which did not provide any gene trees again. Last time also, the other outputs came except the gene trees. this time also I did not get the gene trees and proteinortho provided no results at all.
You can share back those examples if you want help troubleshooting. If this is a problem on the server, we can share that with the administrators to fix what might be going wrong. If you could label what each example represents, or share the “before” and “after” that shows the problem that will make it easier to see where things may need a correction. Thanks! ![]()
This is the history where I uploaded 4 fasta files and 4 gff files after rectification. I did not get any result for gene trees after running Orthofinder using three different parameters. Also, I got no output from Proteinortho.
I am eagerly waiting for your help. I have shared the history with you already.
I started on this last week and my test jobs were running. The data formats seem better now but I wondered if these would work better in a collection, so that is what I was testing. You could try that too, while waiting for feedback from me. I hope to get to it this week, maybe tomorrow! ![]()
Were you able to get the other ortholog tools you are exploring to work? On this same data? I would be interested in comparing between all runs if you want to share those histories as well.
This tool was not outputting the result file – so I do think it is a problem I’m just not sure yet why and if there is something about the inputs we can still adjust. Many tools have little quirks but we can also isolate and describe the odd behavior to the developers, I just need a really solid example first. More soon!
Thank you. I will be eagerly waiting for your results and feedback.
Nevertheless, I uploaded it as a collection in galaxy but while running Orthofinder I couldn’t upload the collection…there I didn’t find the collection option so I uploaded the datasets individually. Could you tell me how to do that?
I just used Orthofinder and proteinortho here. None worked. I am not sure of other ortholog tools. Any idea from your end?
Thank you once again for so much of your constant support.
As a comparison, I created a test history here. Notice that all of the outputs are available using the 4-species example case.
Why your data is not producing the extra output isn’t clear but this is certainly a data-based reason. Maybe by compariing to the example above you’ll be able to spot the why and make a correction.
Also: if you compared all of the genes in the original files to the list of unassigned genes, are there any genes not accounted for? Those would be the set to map out into the other output. If there are none, that would be the “why”.
We can continue with this next week if the accounting for the gene level result is still odd but I wanted to give you some feedback before the weekend. ![]()
Did you find a solution to this? David Emms says there should be an output file called Orthologues.csv, but I don’t see that on Galaxy Europe. I was hoping to use this file against the Orthogroups output to isolate the paralogs.
My test case above has the output you are asking about (I think .. please double check me!)
Screenshot of the dataset for this output file. It is in tsv format (tab separated, easier format when working with other tools) but you can easily convert to csv (comma separated) if you want to download it to do something else.
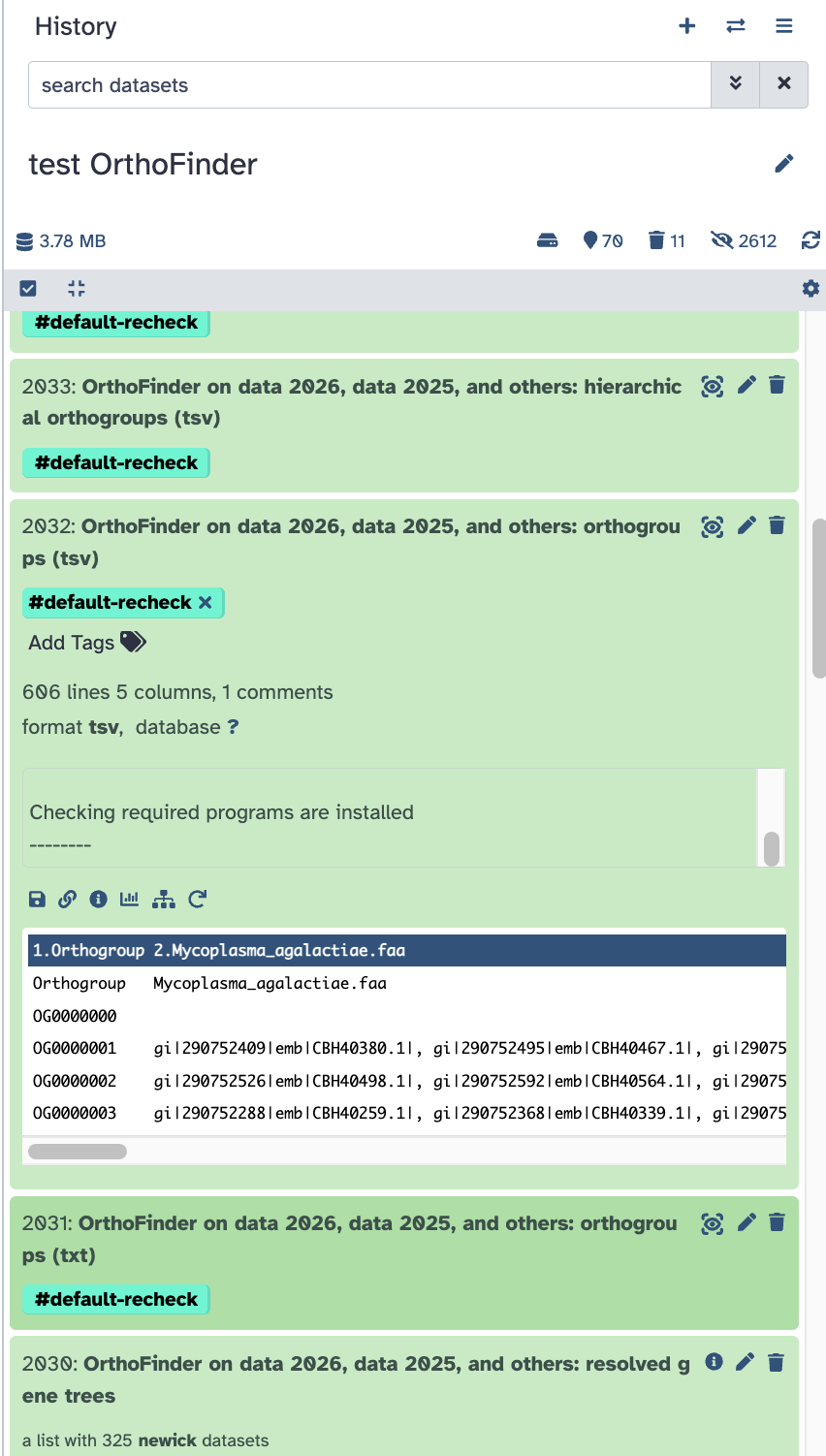
Does this help?
And @Sutrisha_Kundu – did you solve this? My test were somewhat small and used default parameters. We can go back and compare together if you are still stuck and want to share where you are now.
I see orthogroups, but not orthologues. It should be a separate file or set or files.
Hi @jaredbernard From what I understand, the grouping of genes into “Othogroups” is what this tool produces. Then it ranks the relationships – each group has an associated tree. See the Resolved Gene Trees.
Am I misunderstanding? I am comparing to the most current version of the tool. This was updated in Galaxy around the time the underlying tool was last updated (about 4 years ago).
- Developer → OrthoFinder/README.md at master · davidemms/OrthoFinder · GitHub
- Source → https://github.com/davidemms/OrthoFinder
- Galaxy wrapper dev histroy → History for tools/orthofinder - galaxyproject/tools-iuc · GitHub
I don’t think there is an output with just a two column file type of format in earlier versions but in the latest version, this is represented in a tree format and includes duplications within the same species, too.
Quote from the tool form for more context. I do see that this tool has evolved. Earlier versions seem to have had different output types? Let us know what you think. If there is a problem, we can get it corrected but I don’t see it yet.
Help
OrthoFinder OnlyGroups
Full readme at https://github.com/davidemms/OrthoFinder/blob/master/README.md Summary sketch at https://github.com/davidemms/OrthoFinder/blob/master/OrthoFinder-manual.pdf
OrthoFinder is a fast, accurate and comprehensive analysis tool for comparative genomics. It finds orthologues and orthogroups infers gene trees for all orthogroups and infers a rooted species tree for the species being analysed. OrthoFinder also provides comprehensive statistics for comparative genomic analyses. OrthoFinder is simple to use and all you need to run it is a set of protein sequence files (one per species) in FASTA format (Emms, D.M. and Kelly, S., 2015).
This galaxy tool implements the first part of the Orthofinder program, e.g. the clustering of orthogroups of genes.
Thanks for getting back to me, @jennaj. Maybe I’m confused, or maybe the results I’m looking for are included in another output.
On the OrthoFinder github account, either David Emms’s or the new one, you can see the output called “OrthologuesStats” files in the Comparative Genomics Statistics Directory (link here). According to David Emms, a person could contrast those ortholog files against the orthogroup files to get the paralogs. But maybe this is somehow in one of the Galaxy outputs and I missed it.