I uploaded a paired-end SRR file using fastq-dump. The filetype says it is a fastqsanger.gz but when I try to run Trimmomatic or Trim Galore, they both say they cannot find any fastqsanger.gz datasets.
Is this not the correct way to do this?
Hi @ajnaram!
That is the correct way.
Both of the fastqsanger.gz datasets are green in color? And this datatype was assigned by the tool used (not assigned/reassigned by you?)
If the data upload was successful, check to make sure you are launching the tool forms with the target datasets in the currently active history? Click on the “refresh history” icon to confirm:

If the history content changes, then go to your saved histories, switch to the history with your data in it, and try again.
One last thing to double check (would be rare now) is if the data is in a related format of fastqcssanger.gz. (Note the “cs” in the name). Sometimes people assign this datatype by accident or the data uploaded really is in the color space format. The tool Fastq Groomer can be used to transform to fastqsanger (if actually needed – use the tool FastQC to check).
FAQs that might help (see the first three): https://galaxyproject.org/support/#getting-inputs-right
Let us know how that goes and we can follow up from there. Please confirm that you are working at Galaxy Main https://usegalaxy.org as indicated by the tags added to the post.
Yes the datasets are green and the datatype was assigned by the upload tool.
My correct history was active.
I didn’t have any cs files but I ran through fastq groomer just in case. Still the same problem.
The same error occurred in the .eu version, but I initially saw it in the .org version.
Thanks!
Could you please post a screenshot of one of the unrecognized fastq datasets? Expand it first (click on the dataset “name”). We might be able to identify the problem from that view.
If that doesn’t work, I’ll need to look at your account at Galaxy Main https://usegalaxy.org as an admin.
- If your registered email here at this site is the same as used for your account at the “org” server, just post back the name of the history and a few of the problematic dataset numbers.
- If the email addresses are different, please send me a direct message here with your email address with the same history/dataset info. You don’t need to post your email address publicly.
But let’s try a posted screenshot first. This Q&A might help others, later on, should they run into the same type of issue.


Hi - I couldn’t find your account at https://usegalaxy.org.
That said, this looks like an interlaced fastq dataset in compressed format. It should be recognized by tools based on the assigned datatype “fastqsanger.gz”. But you might run into problems after that.
Most tools will expect that paired end data is in two distinct datasets – one representing the forward reads, one representing the reverse reads. This includes Trimmomatic and Trim Galore!.
Run the tool Fastq De-interlacer to split up your current data, then see if the tools find your inputs correctly.
FAQ: https://galaxyproject.org/support/#getting-inputs-right
Thank you for the help!
I’ll try the de-interlacer though it was buggy for me the last times I tried it (reported bug).
Hum, ok, if failed there is probably something wrong with the sequence identifiers. The tool requires some specific formatting to split the reads.
The FAQ has an alternative method that is custom, based on your identifiers, whatever they may be. There are also a few other ways to do this – let us know if continue to have problems and I’ll look directly at your data and help. Not every bug report has the data examined in granular detail (for obvious reasons) … but we try  and can help, especially if you get completely stuck at some step.
and can help, especially if you get completely stuck at some step.
Hi @jennaj, I’m having a similar problem (hence replying here and not posting a new question). On the main instance (usegalaxy.org) I have a history called “RNA seq of Cdub and Caur” (my user there is registered under this e-mail). I successfully imported a FASTQ sequence from NCBI (SRR771366) with fasterq-dump (item #2 in the history). However, RNA Star does not recognize it as a valid FASTQ input. I uncompressed it (item #20), and that version was recognized. It seems to me that FASTQs imported this way should definitely be recognized - did I do something wrong?
The tool “Faster Download and Extract Reads in FASTQ format from NCBI SRA” outputs data into collections by default.
This means that you need to look for a collection when selecting the input on tool forms. Make sure that the “single-end” or “paired-end” is a match between the form settings and your actual data.
Form option set to search for appropriate “single” datasets. In this example, there are not any in the current history.

Form option set to search for appropriate “collection” datasets. In this example, there is one in the current history.

(facepalm…) Thank you! Is there a way to rename the collections? It seems they have no “Edit Attributes” button, and as for the data contained within, while I can rename it, the new names are not updated in the history (but are updated in the attributes panel).
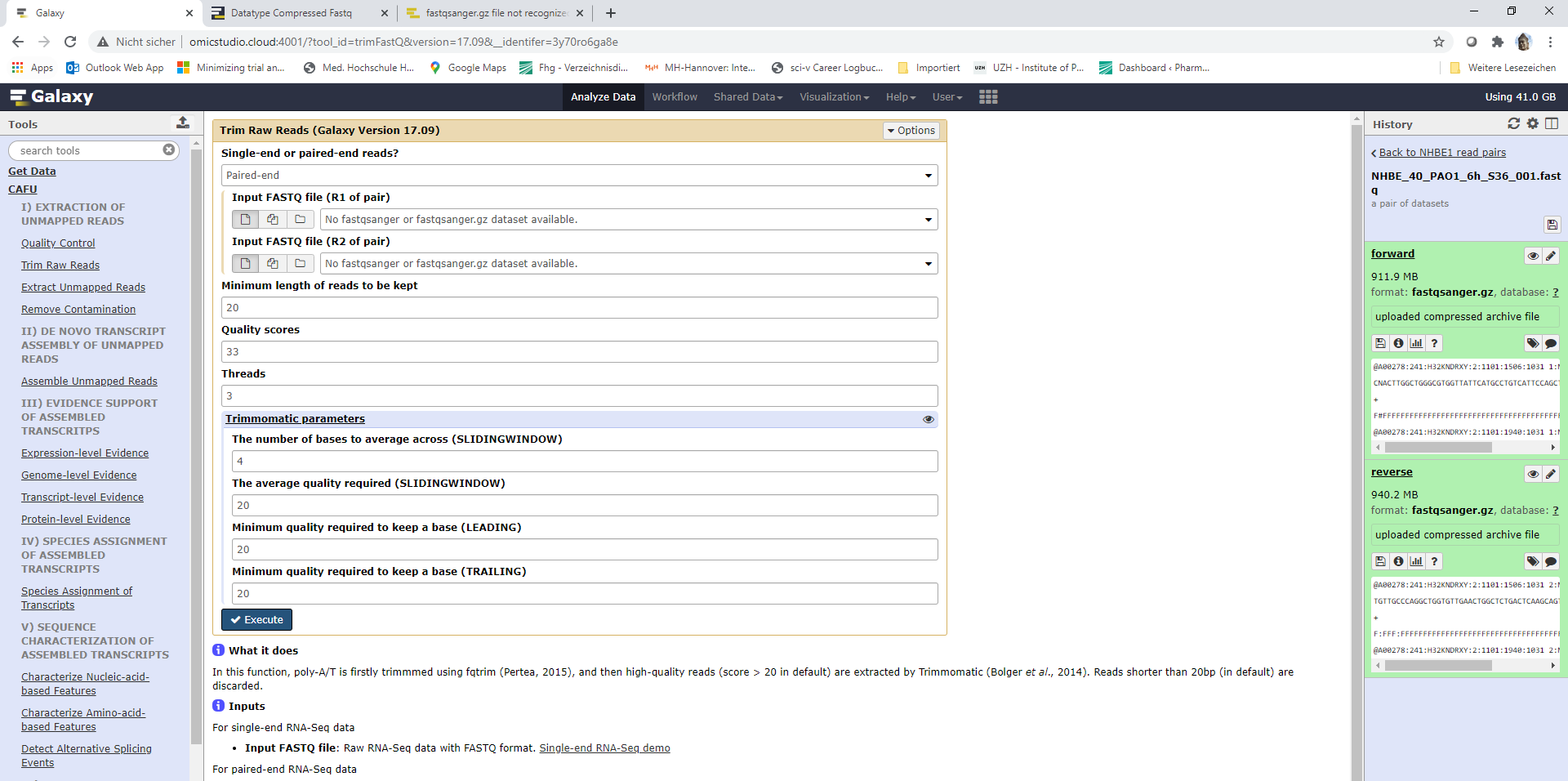
Hi, I’m running into similar problems here, using the CAFU framework. I uploaded dual RNA-seq data in fastq.gz format. FastQC ran without problems.
However, when I want to use trimmomatic, it doesn’t recognize my datasets. My datasets are in a list of paired reads. I already manually changed the datatype of each dataset to fastqsanger.gz but it still does not work. I read the FAQ on Datatype Compressed Fastq but could not figure out the mistake.
Any ideas, what I might be doing wrong? I attached a screenshot.
Thanks for your help!
{kind=link}