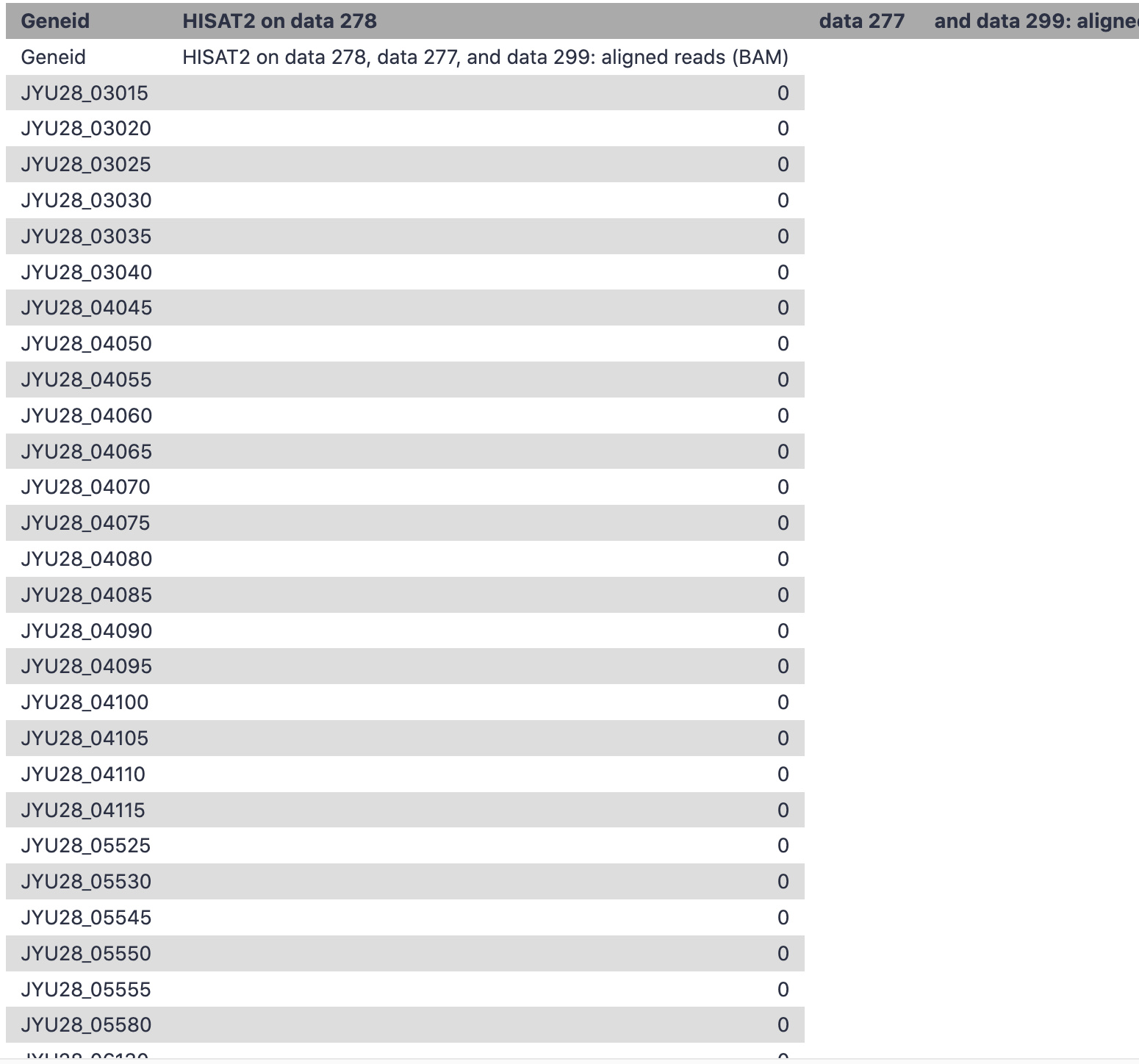
I am trying to align my RNA-seq reads (Bacteria), using HISAT2 to a reference genome (assembly.fna). Then,to measure the expression in the resulted bam.file, I used (featurecounts) tool, using the (gtf) annotation file. However, although both fna/gtf files belong to the same strain tested, I got this empty report;
Your help much appreciated
No, not the file names, the sequence/contig names.
Click on Preview (eye) icon of the reference genome (FASTA) file. Check the sequence name in the first line after ‘>’. Most tools consider a text string before the first space character as a name, and the rest is a comment.
Click on Preview icon of the gene annotation file (GTF). Check the name in the first column (seqname). Sometimes the annotation file has a header. Header lines start with #. Ignore the header lines.
Both files should have identical sequence name.
This is assuming the assembly contain a single sequence. If you click on name of FASTA file, Galaxy shows number of sequences present in the file. Some assemblies may have more than one. If it is the case, filter the genome assembly (FASTA) file using something like ^> (select lines staring with ‘>’) and check that the annotation file uses the same sequence names.
Kind regards,
Igor
Alternatively, you can compare sequence names in the BAM file (SQ line(s)) and the gene annotation file. I assumed you use reference genome from history, hence mentioned FASTA file.
So, it is not the reference data.
featureCounts summary indicates majority of reads are mapped outside of the annotated genes. Sometimes RNA-Seq libraries are extremely enriched with rRNA sequences. Run Samtools idxstats on BAM. Do you see reads mapped to all contigs? Do you see a contig with unusually high number of aligned reads, something like >90% of reads?
Hi @Seraph1,
the reads are mapped to many contigs. This is a good sign.
If you share the history I can have a look. As you got no counts for genes, mismatch between the genome and the gene annotation is the prime suspect.
History sharing: History menu (cog wheel icon at the top right corner of the History panel) > Share or Publish > Create a link and paste in in reply.
If you have many samples, you can copy files for one sample (reference genome, gene annotation, BAM and featureCounts outputs) and share only one sample.
Kind regards,
Igor
Hi @Seraph1,
output of featureCounts #14 has counts for 157 genes, so it does count reads against some genes. The small number of genes is an unintended consequence of the gene annotation. Tools such as featureCounts and htseq-count count reads against a feature in 3d column of GTF file and aggregate the results using an attribute from the last column. For example, feature in the 3d column can be ‘exon’, and attribute is ‘gene_id’. The tool counts reads against axons and returns count for a value (gene ID) in ‘gene_id’. Your GTF file does not have ‘exon’ feature for some genes. Filter the GTF file on column 3 using Filter data on any column using simple expressions with c3==‘exon’. You’ll see 157 records. As bacterial genes are not spliced, the featureCounts estimated read counts for genes with available ‘exon’ feature. You can select a feature used by featurCounts for counting in Advanced options. If you set it to CDS, featureCounts returns 5092 lines. With CDS you’ll miss some ncRNAs. Try different features, examine the annotation, and select a feature most appropriate for purpose of your analysis.
You can unshare the history if you don’t have any other questions: History menu > Share or Publish > Unshare it.
Kind regards,
Igor
I have the same issue, but I am dealing with the chicken genome.
Alignment with HISAT2 worked perfectly fine and featureCounts runs without an error message, but I have 0.0% assigned reads throughput my samples. I tried it now with several features and options possible, still 0.0% assigned reads.
I can also exclude bad samples quality because it worked before.
Hi @Mila
do you use paired end data? If yes, in featureCounts job setup check Options for paired end reads and enable Count fragments instead of reads.
Hope that helps.
Kind regards,
Igor
Hi Mila,
two common causes: different contig/chromosome names in the reference genome and gene annotation file and different or missing feature (column 3 in the annotation file) and/or attribute used for aggregation of counts. Compare values in featureCounts ’ advanced settings and the annotation file.
If there is nothing suspicious, copy one BAM file and gene annotation in a new history and share it with me, for example, post URL here.
Kind regards,
Igor
Hi @igor, I am facing a similar problem with my featurecount where it says it cannot find attribute in the column. I am using my gene annotation file as the bacteria I am working with does not have a built-in gene annotation
Could you please go through the screenshots and help me with this.
I am sorry for cut and paste, but:
Tools such as featureCounts and htseq-count count reads against a feature in 3d column of GTF file and aggregate the results using an attribute from the last column. For example, feature in the 3d column can be ‘exon’, and attribute is ‘gene_id’. The tool counts reads against axons and returns count for a value (gene ID) in ‘gene_id’. Your GTF file does not have ‘exon’ feature
Make sure that in setup of featureCounts job you use feature type and gene identifier existing in the annotation file.
Hi @parvati.iyer
Can you get the same annotation in gtf format? As I said, I see less issues with gtf. If not, convert gff3 to gtf using gffread.
You don’t need “=” in gene identifier (in job setup).
Kind regards,
Igor