Hi @Kunyuan_Tian,
I think this is the tool that you require: Select. You are right, it doesn’t show up when typing Select, but the search bar will be improved quite a lot in the next release.
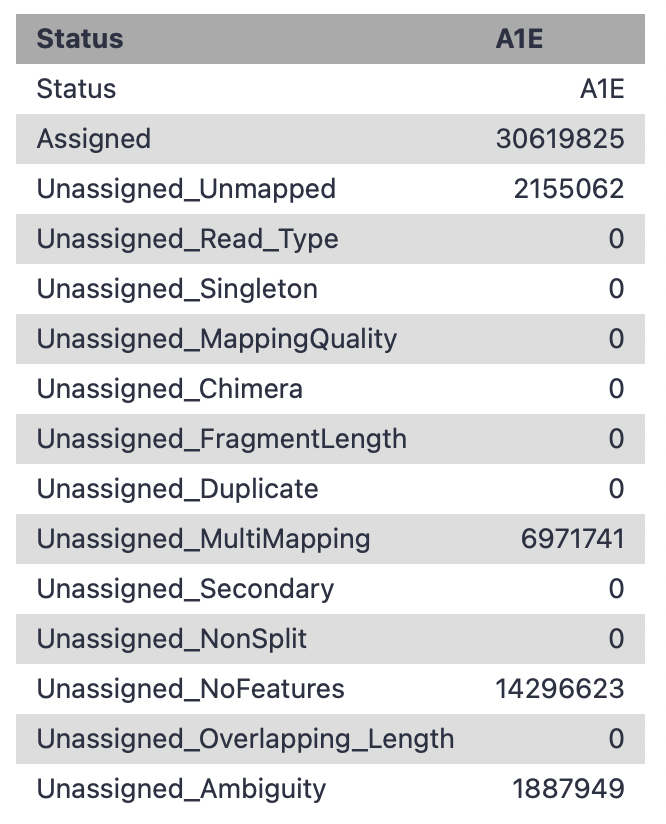
What would likely be the reasons for high number of noFeature reads? So far to my knowledge, there’s less than 0.1% adaptor content after Cutadapt, the RNA STAR alignment was decent (90% uniquely mapped reads). Library is unstranded as I tested with infer experienment and had 47% for both forward and reverse. I used GRCm39 for alignment and uploaded GRCm39 annotation from Genecode. Any advice on how to unravel the high no_feature read would be really appreciated!
The scientific and technical reasons other people have run into are in several other Q&A topics at the author’s support forum Bioconductor Forum and more are here just about Galaxy. There will be some overlap. Please have a look first then share the more details about your analysis if you still need more help (or an R → Galaxy tool form translation).
Some basic checks include: Did you map against the built-in index mm39 (at usegalaxy.eu – the UCSC version of GRCm39)? Are you certain that the genome annotation is in GTF format (Gencode also hosts GFF3 format)? Gencode also host multiple annotation subsets – maybe you need to try a different one? You could also backup – did the original and post-trimming read QA/QC steps reveal anything special about the content? Is the data public or your own?
Your reads are mapped but not overlapping with annotated regions. Opening the BAMs and your GTF in a tool like IGV might reveal what is scientifically going on after any remaining technical issues are resolved.