Hi @rcaudle (and others that may run into this issue!)
Thank you for sharing the examples! There are two issues going on. One with the way the cat tools work at the ORG server, and one with the cluster failure handling at the ORG server.
First: how Text Manipulation tools handle compressed datatypes
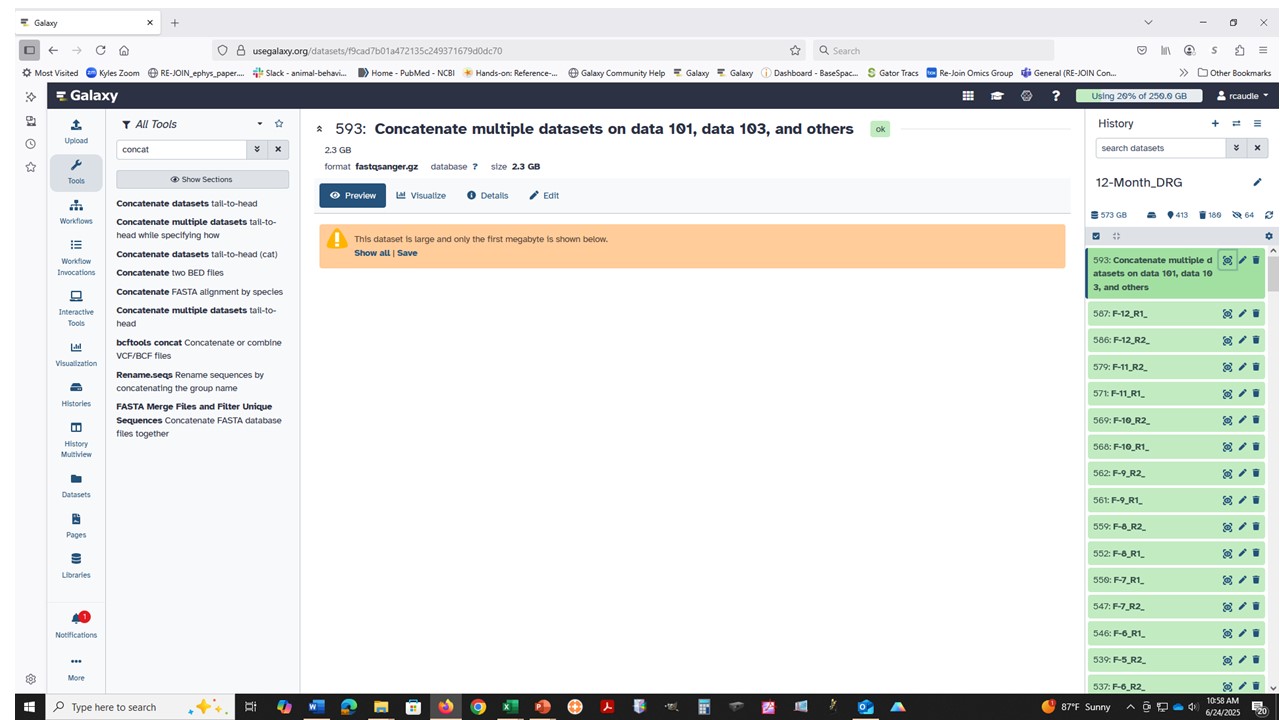
At UseGalaxy.org (may be different at other servers): The concatenate tools might work on compressed fastq data but please don’t rely on that. Most of the forms have a version of this type of warning.
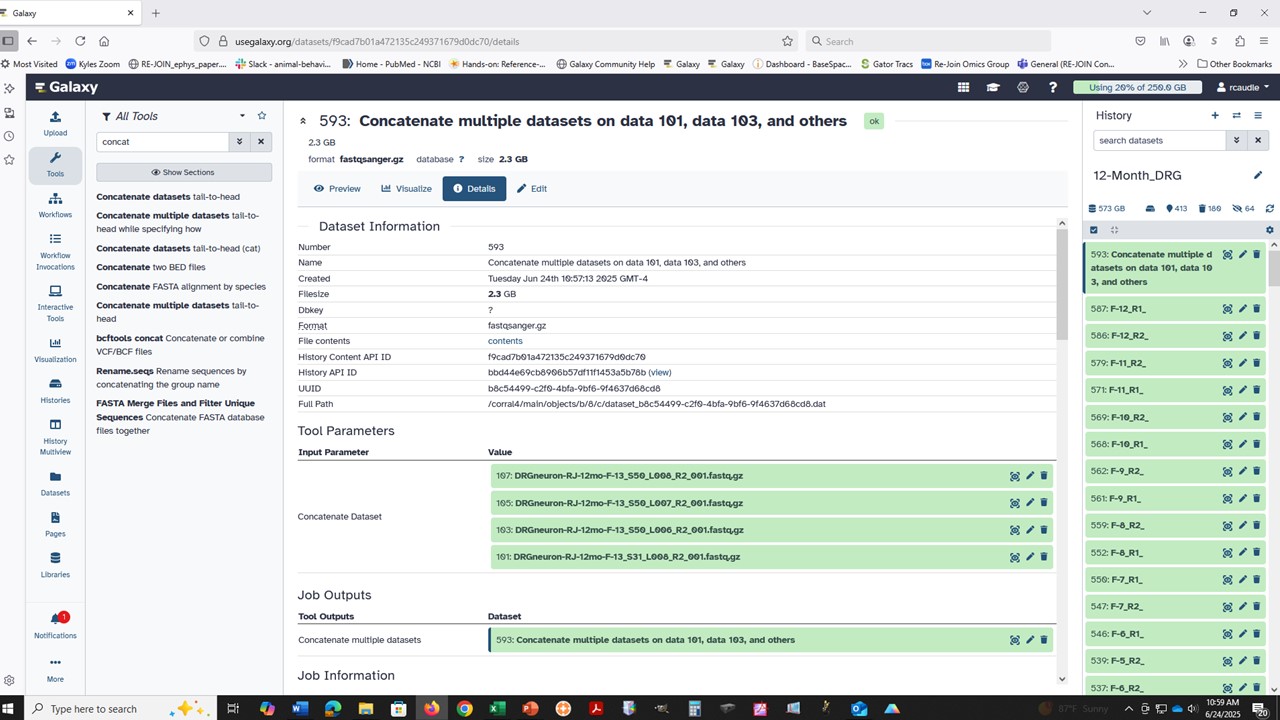
This tool does not check if the datasets being concatenated are in the same format.
It means that the usual datatype handling/checks that tools perform are not “required” to be handled by this specific tool (not called out in the job environment, which is important for choosing suitable clusters to use). Most of the Text Manipulation tools work the same way. The server, and cluster nodes involved, might be able to handle data that isn’t in a plain text format, but it isn’t an explicit requirement. This might explain the inconsistent results you are seeing.
In short, these tools behave the same way as a command-line utility. For cat, the tool simply stacks the data files all on top of each other. No smart datatype filtering is applied in the job configuration set up on (on purpose) to allow for complex or mixed data file types to process. This gives some flexibility when needed, but can be confusing!
Presenting the cat tools as just cat tools, and leaving the file handling up to the user, is how to approach the usage. The solution is to add in the uncompress/recompress steps in to your workflow. Most of the other Text Manipulation tools work this same way: plain text works most reliably across servers and clusters, and you might lose your datatype and need to reassign it!
So, while some cluster node might have the required transformation support for “compressed to uncompressed to cat to re-compressed” in the job environment, that shoudn’t be replied on. You’ll need to make sure the input data is in a plain text format before using one of the cat functions, and that includes the Collapse Collection function (which again, might work on compressed but often will not).
How I would suggest doing this
I would put it all into a sub-workflow to recall whenever needed. This would allow you to split off the multiple-temporary file versions too, since this is usually uninteresting data to save.
 https://usegalaxy.org/u/jen-galaxyproject/h/example-cat-for-compressed-fastq-1
https://usegalaxy.org/u/jen-galaxyproject/h/example-cat-for-compressed-fastq-1
-
 Main workflow (creating the input for the subworkflow): Group the files into a temporary collection (Collection Builder or split off the files you want from the original collection(s) into a new slice with one of the Collection Operations tools/functions). This collection will not increase your storage usage since these are identical clones of pre-existing elements.
Main workflow (creating the input for the subworkflow): Group the files into a temporary collection (Collection Builder or split off the files you want from the original collection(s) into a new slice with one of the Collection Operations tools/functions). This collection will not increase your storage usage since these are identical clones of pre-existing elements.
-
 Sub-workflow: Uncompress all at once (pencil icon → convert or for a workflow Convert compressed file to uncompressed).
Sub-workflow: Uncompress all at once (pencil icon → convert or for a workflow Convert compressed file to uncompressed).
-
Sub-workflow: Collapse Collection (or any other of the Cat tools that you like)
-
 Sub-workflow (creating the output to send back to the main workflow): Recompress all at once (pencil icon → convert or for a workflow Convert uncompressed file to compressed. These will be new elements, and will increase your storage usage, the same as if Step 2, 3, 4 were a single tool.
Sub-workflow (creating the output to send back to the main workflow): Recompress all at once (pencil icon → convert or for a workflow Convert uncompressed file to compressed. These will be new elements, and will increase your storage usage, the same as if Step 2, 3, 4 were a single tool.
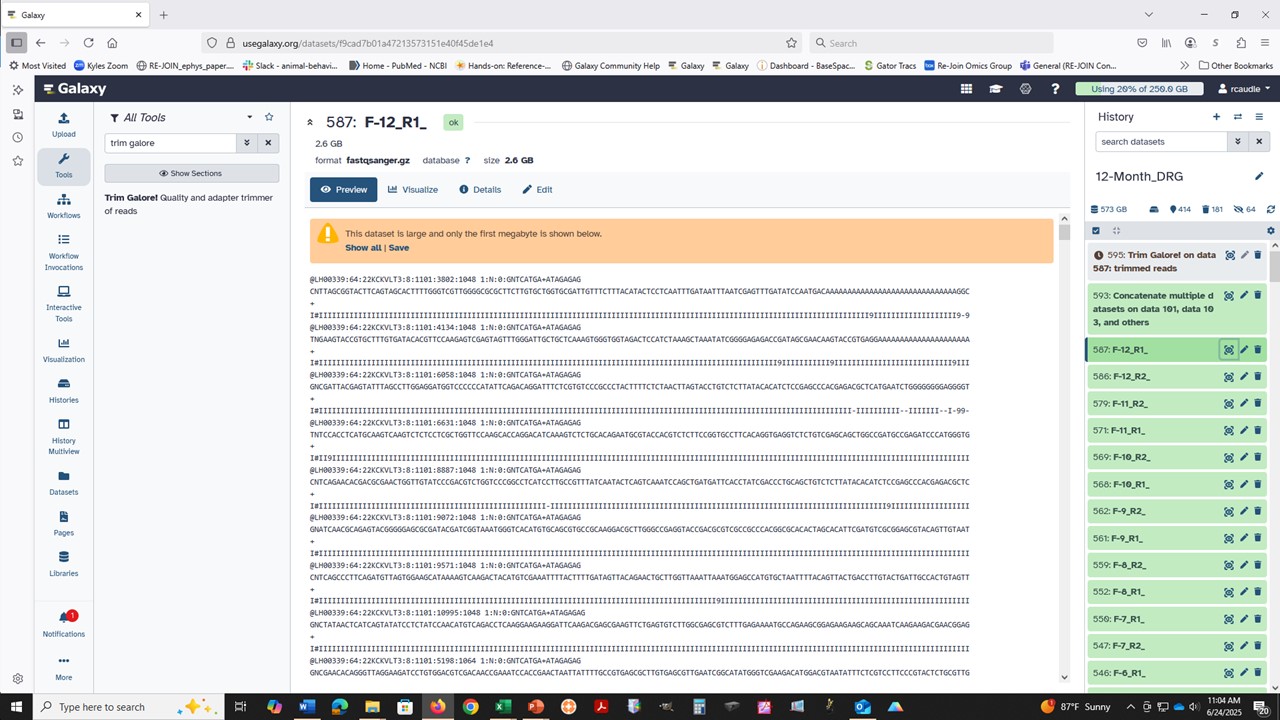
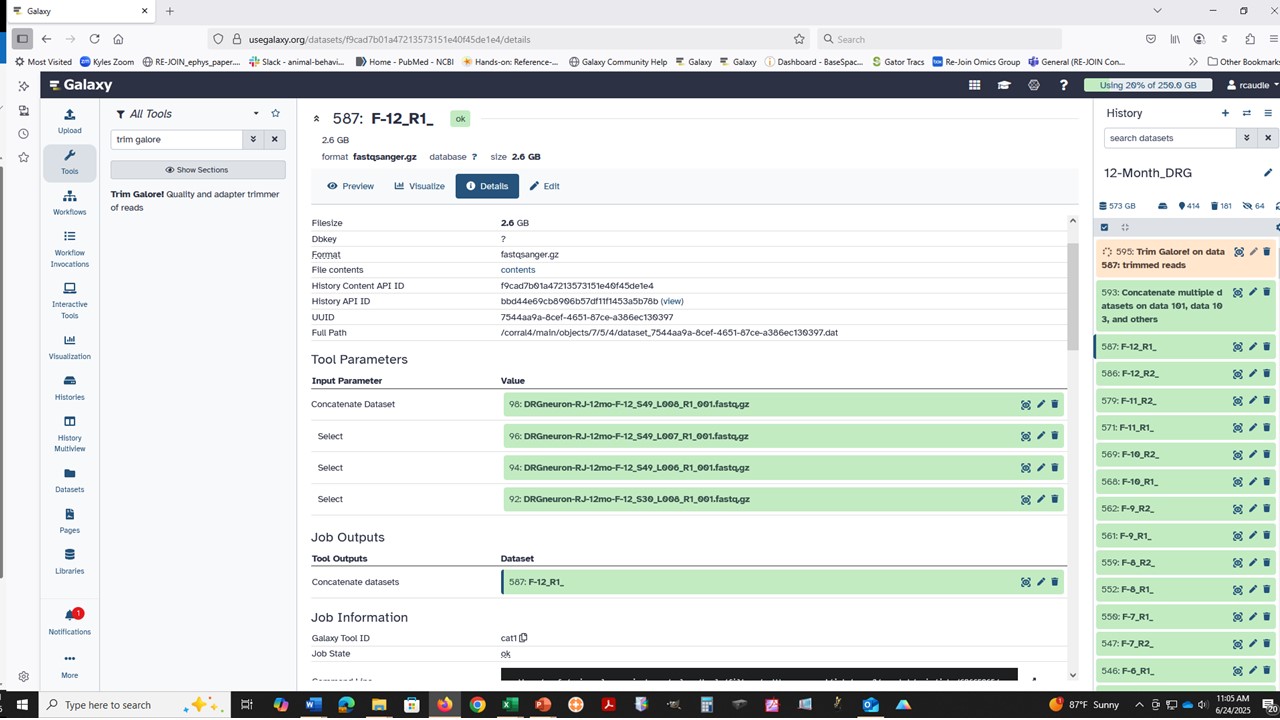
Second, there are some (known) issues with the clusters, which is why these empty result jobs are labeled as successful (green) and the job logs are not reporting the actual issue (it will just report that it can’t read the data, not any details since it is only looking for data eg any plain text, not parsing around a specific datatype!).
We have been working on this for the last two weeks, and that work should start to roll out to more tools over the next few weeks. Until then, failed jobs may not look failed in the expected way (red, with informative logs) and may instead be green, empty, with cryptic format logs (direct from the cluster, not parsed nicely). The issue will usually be due to unexpected content in the inputs – but please ask about anything that is not clear and we’ll help to confirm.
Hope this helps!  Help for how the sub-workflow creation was streamlined in the last release can be found here → 25.0 Galaxy Release (June 2025) — Galaxy Project 25.0.2.dev0 documentation
Help for how the sub-workflow creation was streamlined in the last release can be found here → 25.0 Galaxy Release (June 2025) — Galaxy Project 25.0.2.dev0 documentation
Related Q&A Tool for merging 2x single-read illumina sequencing files (fastq) into one?