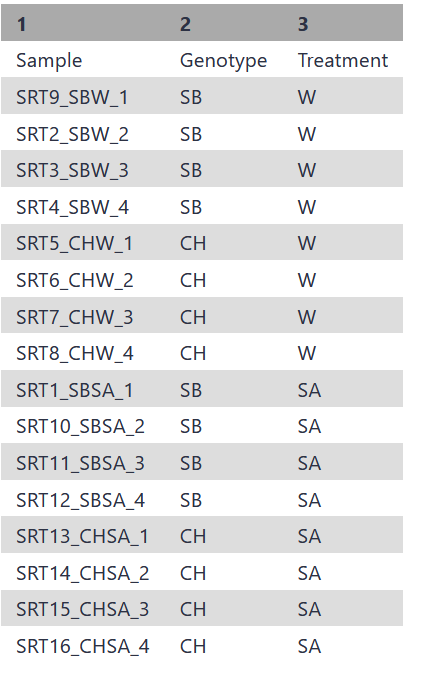
Yes, this sounds like a technical issue… maybe sort order? Try sorting that 2nd column “Genetype” so that the CH rows are all together and first with the order of “Treatment” rows with W first, then SA (repeating as appropriate). One done – and you have a specific order of “Sample” rows, make sure the count files order those samples in the same order.
If you are not sure how to rearrange data – this tutorial covers common tools: Data Manipulation Olympics
If that is not enough, please post back more screenshots of the tool form, other inputs, and job details. Or, you can post a shared history link, how-to: faqs/galaxy/#troubleshooting-errors
and have also reordered the samples in the counts tables to match the factor file order.
However unfortunately, tried to run edgeR once more but still get the same error message of SA not being found.
I have a sneaky feeling that the error could be coming from me creating the factor file in excel and then uploading it as a txt into galaxy, as when I view the TXT(tab delimited) on my laptop, it looks quite messed up
It might be Ok – but you could use this tool to cleanup the delimiters: Convert delimiters to TAB. You could run that any other text files you manipulated.
For an example, this tutorial uses Limma and involves two contrasts. The methods were simplified a bit from what you are doing (uses merged group labels) but maybe is a useful reference anyway. 2: RNA-seq counts to genes