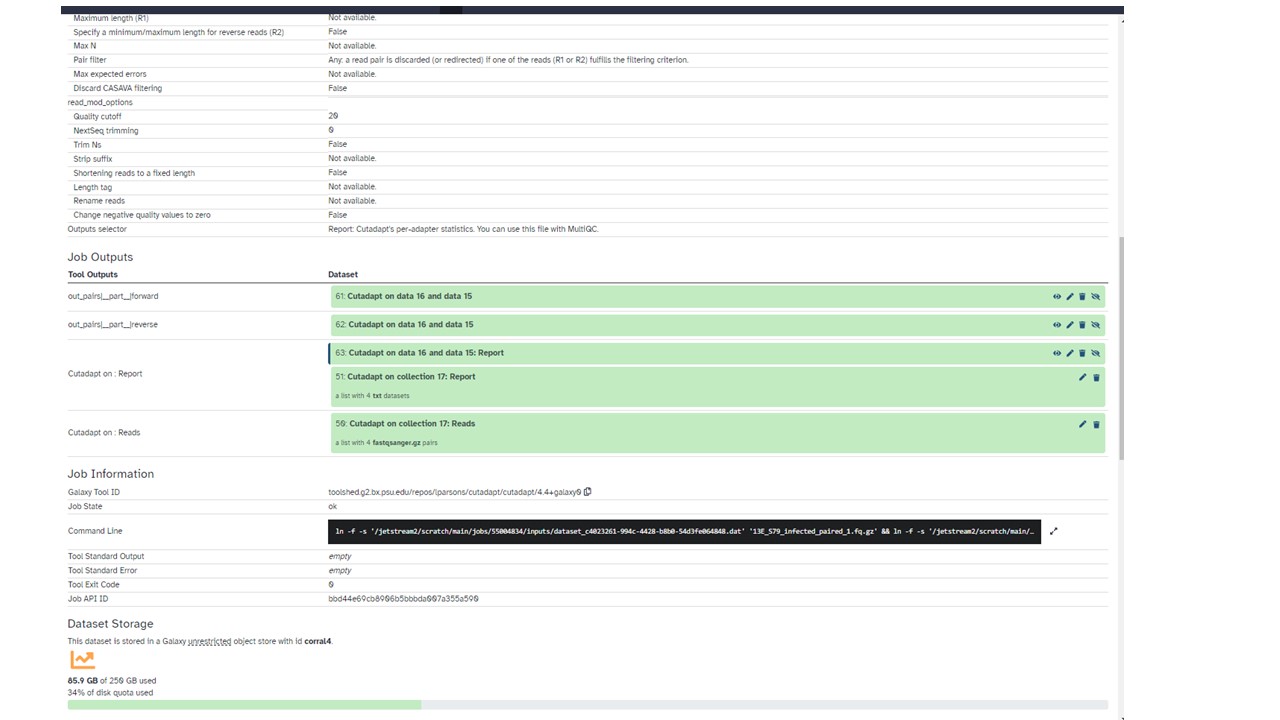
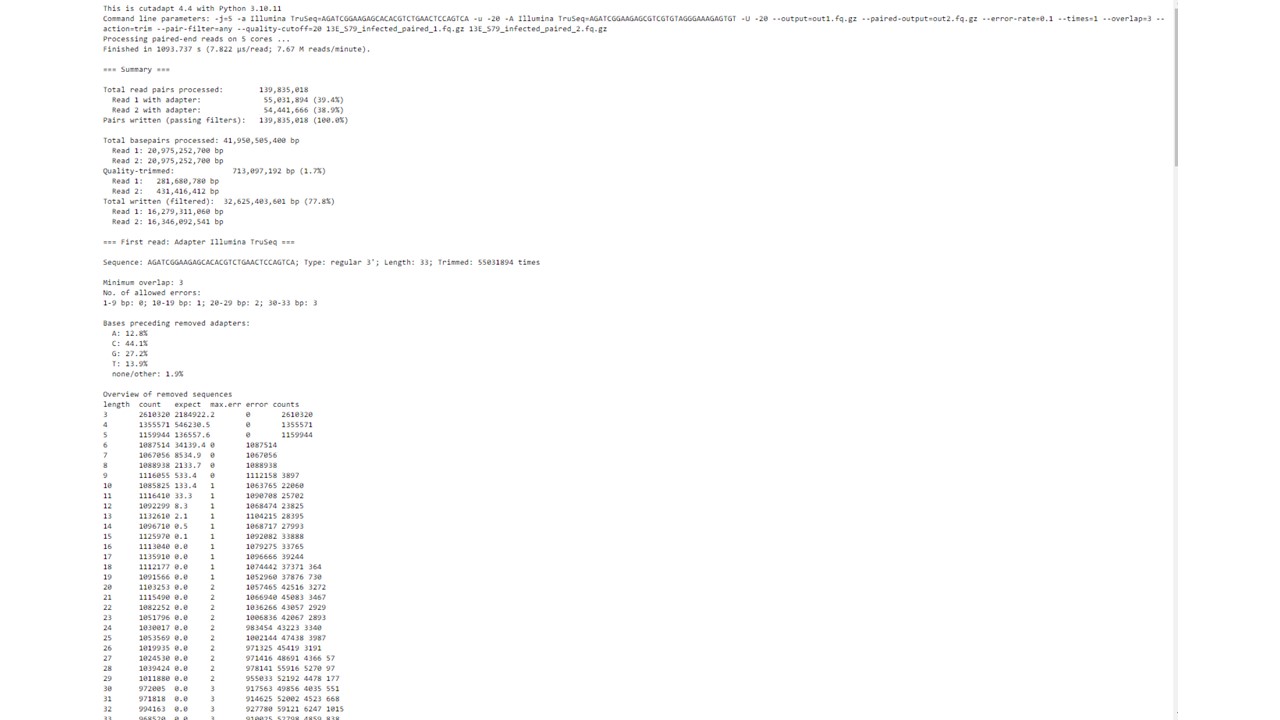
Do the job logs have any more information? The banner at this website explains where to find those, or if you already cleared that away, see this → How to get faster help with your question
Thanks for your response.
I am still having trouble with the mapping and trying to figure out where I went wrong with cutadapt.
So , I still need help solving my problem.
I have attached additional information of my cutadapt and mapping to this message., hopingfully it shows something.
If the QA isn’t what you expected (end up with empty files), then you’ll need to adjust that tool’s parameters (trial and error, and tutorials have examples). You could also try Fastp instead.
If the STAR job fails again, either link back the job logs from that run, or post back screenshots of the logs. The stderr and stdout logs – whole thing since where the message is can different depending on where the failure happened. Also post a screenshot of the annotation file (just the “peek view” of the first 4-5 lines is enough – click on the dataset on that job information view and it will expand a bit).
You are asking STAR to use “feature = exon” in the parameters – that is in the third column of the GTF, and should be in there if sourced directly from the public web links, so I don’t think that part is a problem offhand. This is why I thought the reads were the problem. But you could also run STAR without annotation to see what happens. It could be a clue if it either works or doesn’t work in your testing matrix.
To help quicker next time, sharing the screenshots along with the history share link will let me review the other details without having to ask. You can always unshare once we are done. I’d explain what I looked at. Your choice
If the QA isn’t what you expected (end up with empty files), then you’ll need to adjust that tool’s parameters (trial and error, and tutorials have examples). You could also try Fastp instead.
If the STAR job fails again, either link back the job logs from that run, or post back screenshots of the logs. The stderr and stdout logs – whole thing since where the message is can different depending on where the failure happened. Also post a screenshot of the annotation file (just the “peek view” of the first 4-5 lines is enough – click on the dataset on that job information view and it will expand a bit).
You are asking STAR to use “feature = exon” in the parameters – that is in the third column of the GTF, and should be in there if sourced directly from the public web links, so I don’t think that part is a problem offhand. This is why I thought the reads were the problem. But you could also run STAR without annotation to see what happens. It could be a clue if it either works or doesn’t work in your testing matrix.
To help quicker next time, sharing the screenshots along with the history share link will let me review the other details without having to ask. You can always unshare once we are done. I’d explain what I looked at. Your choice
The second FastQC run didn’t have the input collection flattened before the run, so you only have a report for one end of each pair.
Here is the history I am working it. A copy of yours with a few tests still running. Galaxy
Exact rerun of the failed pair
Cleaned up GTF, then used that with the failed pair in a rerun
FastQC/MultiQC plus Fastq info on the pair (pre-QA) just to show steps
FastQC/MultiQC run on the post-Cutadapt reads (flattened)
One of those should let us know what is going on. I wait in the queue just like everyone else … so you might see the results before I will! Hopefully helps!