I have my datasets from bulk RNA Sequencing. I tried to process my datasets, it’s mouse, using the workflow from Galaxy tutorial. But after the trimming and going in to the alignment with RNA STAR, it gives me lots of error. Like:
Aug 17 10:25:31 … started STAR run
Aug 17 10:25:31 … loading genome
Aug 17 10:33:47 … started 1st pass mapping
EXITING because of FATAL ERROR in reads input: quality string length is not equal to sequence length @A00643:482:HYYN7DSXY:1:1101:2
and error on gtf file
Aug 17 11:17:05 … started STAR run
Aug 17 11:17:05 … loading genome
Aug 17 11:21:48 … processing annotations GTF
Fatal INPUT FILE error, no valid exon lines in the GTF file: /data/dnb06/galaxy_db/files/9/3/e/dataset_93e3f860-b7ec-4bec-8cb2-6d
I’m a noobs in this sequencing analysis stuff. How can I fix the error? I will highly appreciate your help
At least one of your fastq read datasets is truncated, and that starts with the sequence ID in the message. Try uploading that data again, as Galaxy cannot repair uploaded truncated fastq data.
Never map reads unless they pass basic QA checks. The tool may not fail – just produce incomplete results and that are difficult to detect. If the tutorial includes QA/QC steps, this should have been caught during QA and before the alignment step. If the tutorial doesn’t include QA/QC steps, then see this tutorial: Quality Control.
This means that the reference annotation isn’t in a format that the tool can use. See prior Q&A at this forum (via the tag I added to your post), and this FAQ working-with-gff-gft-gtf2-gff3-reference-annotation. GTF will work best with this tool. Make sure any header lines are removed.
@jennaj Thanks a lot for the explanation. That means the trimming result is the culprit, right? Should I skip the trimming and just go straight to alignment?
@jennaj I tried to run my un-trimmed data on RNA STAR, and fixing the gtf file by removing the header but still getting the same error
Oct 10 14:19:56 … started STAR run
Oct 10 14:19:56 … loading genome
Oct 10 14:25:35 … processing annotations GTF
Fatal INPUT FILE error, no valid exon lines in the GTF file: /data/dnb07/galaxy_db/files/a/a/4/dataset_aa45ad6e-56f2-4303-8e43-a1
If the headers are removed, then you can trust the error message more. If the GTF doesn’t contain lines with “exon” in the third column (feature), you’ll need to find a version for your genome species/build that has that annotated data content OR try generating that data with the tool gffread as explained in the FAQ.
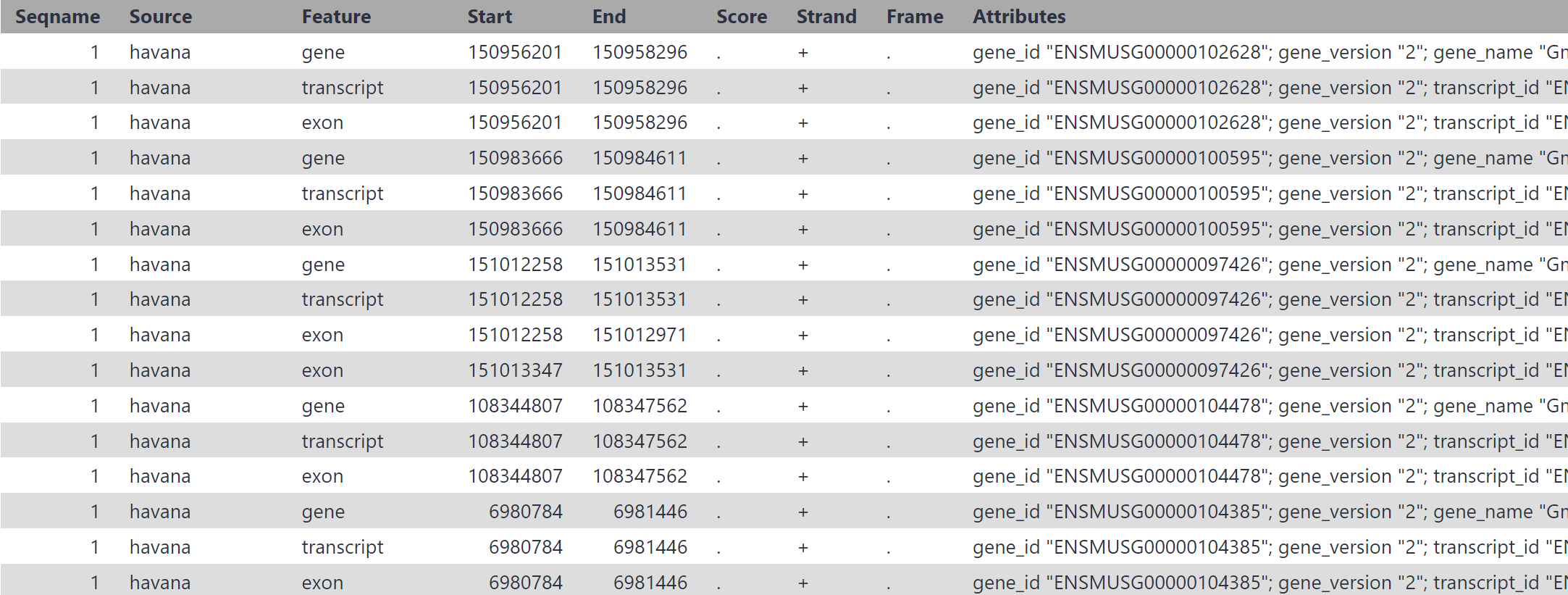
You are mapping against Mouse, correct? If you cannot generate a GTF with the annotation you have now – most public Galaxy servers host UCSC’s version of genome assemblies when available for a species and UCSC hosts the matching annotation GTFs. Those represent annotation from one or more “Gene and Gene Prediction” tracks. Find the files in their Downloads area (avoid the Table Browser – the data is usually too large to extract). Make sure the GTF’s genome build “database” is exactly the same as the reference genome “database” that you are mapping against.
The path is similar for other UCSC genome databases indexed for mapping tools. Copy the URL for the GTF file, paste that into the Upload tool, and leave all settings at default. The GTF will automatically uncompress and be ready to use without other formatting changes.
Your annotation GTF has Ensembl chromosome identifiers like 1. The mouse reference genome index you are mapping against in Galaxy (mm10) was sourced from UCSC, and has UCSC chromosome identifiers like chr1.
Technically, you can also try to convert the chromosome identifiers using this tool → Replace column by values which are defined in a convert file.
This will involve multiple steps, and might not work well.
Instructions are on the tool form – be sure to scroll all the way to the bottom to find where to source a mapping file.
Other related methods are included in this FAQ. All involve complex manipulations without exact help, and none are recommended except in extreme use-cases. I posting this mostly for reference, and not recommending that you try it. mismatched-chromosome-identifiers-and-how-to-avoid-them