I have been trying to use single-cell RNA-Seq alignment and bar code processing software STARsolo on four different samples from the same experiment. However, only one of the samples can be processed - the one with the smallest number of sequences.
Each time I try to run a sample, I get the same basic error:
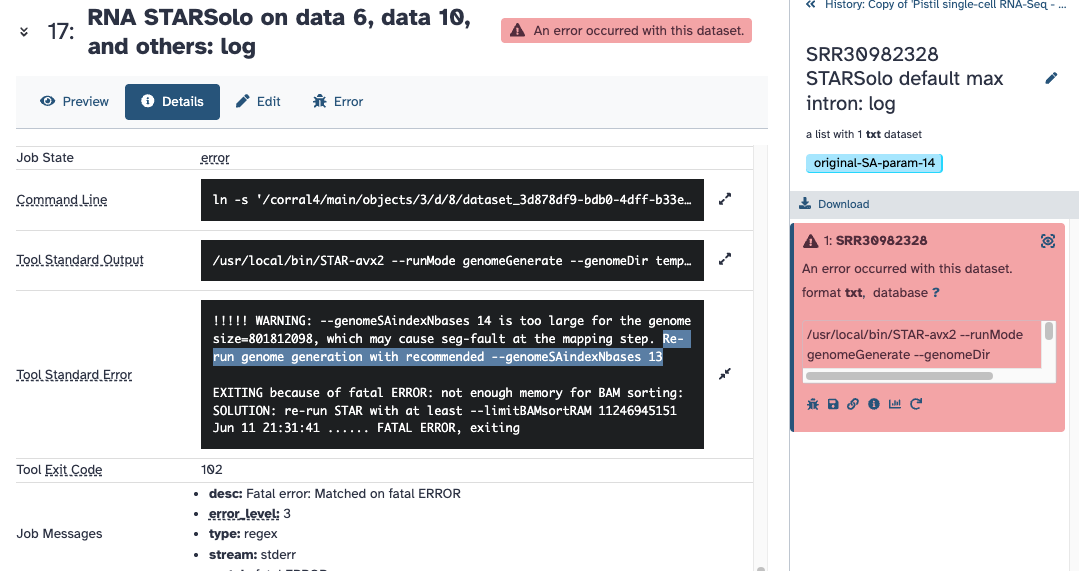
EXITING because of fatal ERROR: not enough memory for BAM sorting:
SOLUTION: re-run STAR with at least --limitBAMsortRAM 11246945151
Jun 11 21:31:41 … FATAL ERROR, exiting
The error messages suggests I can fix this problem by providing option --limitBAMsortRAM 11246945151. However, I don’t see this as a possible option in the tool’s parameter settings in Galaxy.
I made a History public in case someone has time to look at the results and let me know how I can fix the problem.
Here is the link to the History:
Thank you in advance for any help you can provide!
You can try to increase --outBAMsortingBinsN from the default 50 to 100 or even 200, though it will require a large number of open files.
It seems like there a few loci that are very highly expressed, which STAR sorting cannot deal nicely with… So it may be a safer option to use the samtools sort.
Is there a way that I can change --outBAMsortingBinsN?
It looks like some of the exon features on the custom 0 chromosome are not bundled into a transcript feature. I corrected that and started a test to see what happens. All the other data features in the fasta and gtf look great!
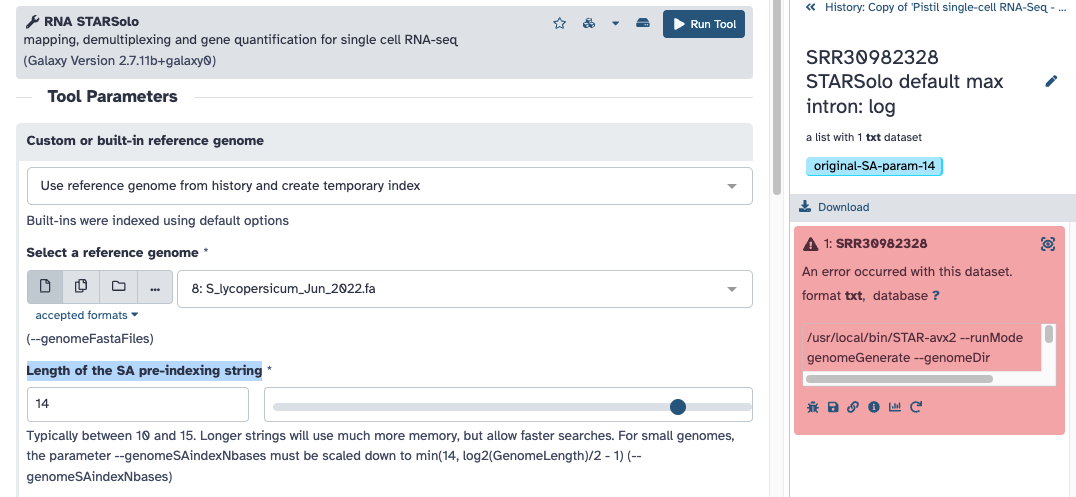
I also adjusted the SA parameter to be 13 in another test (what the logs are reporting to try). Is this what you had trouble finding? Do these screenshots help?
Parameters like this one are set by administrators to fit the clusters being used. These are fixed at the public servers. If the data changes being tested in the other history are not enough, then this job will be too large to process at the ORG server (with the other current parameters) and you can try next at the EU server.
But let’s confirm with the single cell researchers. Maybe there are some settings that can be adjusted for very deep read pileups. I’ve cross posted this over to their chat and they’ll usually get back to us here, but feel free to join there too! You're invited to talk on Matrix
And for @aloraine the samples look fine: completely loaded, contain what I’d expect for single cell reads, and the deeper coverage can be seen a bit in the slice stats.
I’ve leave all that here in case you want to take a look, then I’ll purge my copy on Monday.