Hi everyone, I have uploaded my reference genome and reference transcriptome as a link for import in galaxy, in some cases when I click on the uploaded file in the history, the sequence of nucleotides is displayed, but in some cases the ascii code is displayed and galaxy has noticed that:
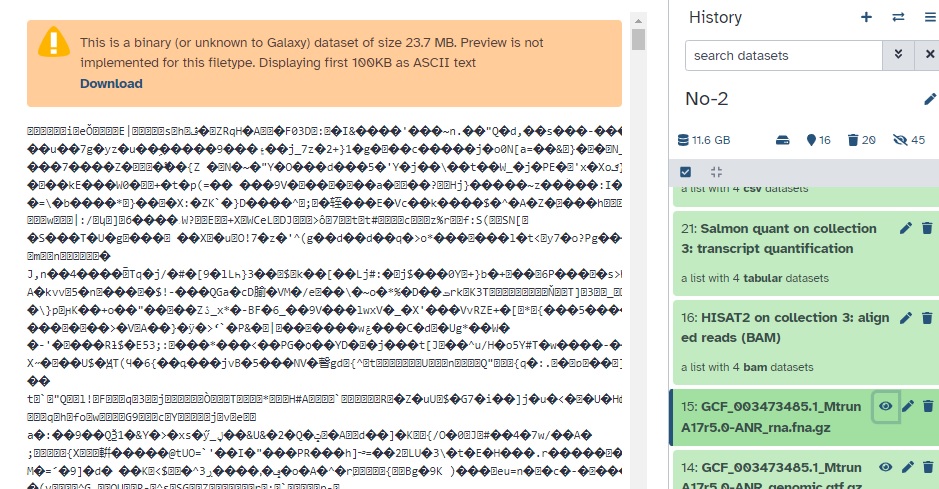
"“This is a binary (or unknown to Galaxy) dataset of size 23.7 MB. Preview is not implemented for this filetype. Displaying first 100KB as ASCII text”
It sounds like Galaxy couldn’t interpret the compression format of some of your files. Or, the wrong compression datatype format was directly assigned in the Upload tool.
Were these public links? You can share those if you want help determining if the compression is something Galaxy can understand. In general, uncompressed and gzip compressed files are best. Let the Upload tool auto-detect the format for any sequence types of data (reads, genome, transcriptome) and any reference data (gtf, gff3). Getting Data into Galaxy
If these were data hosted somewhere else, can you share more about that? From where? What was the original compression format?
In short
It is usually possible to move data between different cloud environments… but that might mean using a specific “share link” from the source.
In any use case: the link must be public for a public Galaxy server to access it correctly.
When working with privacy restricted data, there are other Galaxy options available (one example is AnVIL).
Let’s start there, and you can share more details if you want help troubleshooting this. It is hard to guess more without more details. Thanks!
then I used the links upload----> paste/fetch----> paste links,
in the history I have the files, but when I click on them, they are displayed in ASCII codes
i tried another way, first downloading the files from the databases and then importing them via upload----> local file. This way the sequences from the reference genome and the transcript are correct, but unfortunately it takes longer to load the files,
question 2: When the reference genome and reference transcriptome are displayed as ASCII code, all analyses are performed based on these references!
This means that I have a BAM file from Hisat2 and TPM from the salmon quant, are all of them wrong? how is it possible that GALAXY cannot interpret the references format, but performs all analyses based on these references?
Great, thank you for posting back all the details!
I was able to replicate what you found, and I think all of this is expected.
This is the history where I used the Upload tool to get the data into Galaxy by URL (copy/pasted directly from the source) then used the pencil icon to create an uncompressed version of all three datasets.
How I am interpreting the data → The files are gzip compressed fasta data with an extension like file.fa.gz or file.fna.gz.
General/anywhere
fna specifies nucleotide fasta format
fa is used for both nucleotide and protein fasta format (happens to be nucleotide fasta format for your example data links)
faa is how to clarify protein fasta format
adding on gz to any of the above specifies that the file is gzip compressed
Galaxy
fasta is the datatype for nucleotide or protein fasta format
fasta.gz is the datatype for a gzip compressed version fasta format
Some tools can interpret the compressed version but not all.
Any problems? uncompress (using the pencil icon) and try again. Creating a Custom Genome Build database key is one example that will expect uncompressed fasta.
In the history panel, a “peek” view of the file is displayed for visualization purposes - just the first few lines. Both fasta and fasta.gz data will display in plain text (uncompressed). This is for convenience.
Using the eye icon to visualize the entire file? fasta will display more lines, but fasta.gz will display the compressed ASCII view you found.
I think all of this is expected behavior, or at least the original behavior. For some reason I thought that the peek view wasn’t displaying either! And I though you may have had problems with reference data (gtf or gff3 formats) – those should always automatically uncompress with the Upload tool, and is how tools expect to work with that data format.
Just since it might come up later and might be confusing: the reverse happens for BAM compressed data. The history will not display a peek view, but the eye icon will display an uncompressed SAM view.
So, why? Uncompressing these really large files in a web application takes resources, and can slow down performance. What we have now is a balance of what seemed practical but still useful. But that can change over time!