I am analyzing ChIP-seq data for identifying H3K4me3 epigenetic marks between a Wt and the deletion of my favorite methyltransferase. I have used for the mapping Bowtie2, for Peak calling MACS2 and for generating bigwig files using the bamCoverage tool.
In short, those below are the output files that each tool mentioned above produced:
Bowtie2:
-bam
MACS2 :
-Peaks in tabular format
-narrow Peaks
-summits in BED
bamCoverage:
-bigwig
I would like to construct first a matrix, with the tool computeMatrix(DeepTool), that is going to be used then as an input to the plotHeatmap (DeepTool), to produce a heatmap that shows the corresponding gene-related regions where the H3K4me3 was located in the WT and reduced or vanished in the deletion of my MTase. To do so, I am currently using as inputs for the computeMartix tool the following:
Regions to plot: The summits in BED file that MACS2 produced
Score file: The the corresponding two bigwig files that bamCoverage tool gave for the Wt and the MTase deletion samples.
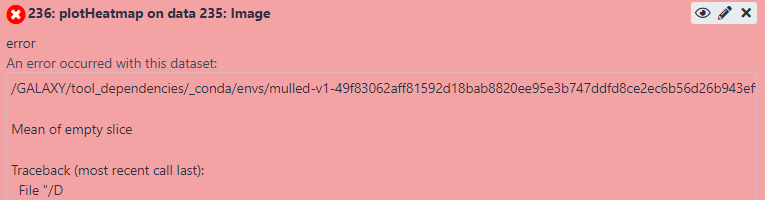
I do get a matrix without any error, but when I am using this matrix as an input to the plotHeatmap tool then I am getting the error below:
I do not understand where the problem is, but I suspect that something with my input matrix is not ok. Thus, maybe I did not use the proper inputs or parameters in the computeMatrix tool… but I do not know what else should I use… I have also searched on the web for similar issues but I was not able to find anything that would help me.
I would highly appreciate any help with that matter,
The tool is reporting that it didn’t find any data where it was asked to look.
Please share more details of your failed job. How the inputs/parameters were set up on the tool forms is usually the best way to start troubleshooting. The more exact the better – so please share this directly in the context of your Galaxy history. Leave all of the upstream inputs/tool runs undeleted, since we might need to backup and review those as well.
Any persistent problems can be reported in a new question for community help. Be sure to provide enough context so others can review the situation exactly and quickly offer advice.
First of all, thank you very much for your prompt response, and my apologies for my delayed reply.
This is how I solved the problem: since the .bed file from MACS2 did not work as an input to the computeMatrix tool, I thought that maybe another file produced by MACS2 (associated with the identified peaks of my specific set of analysis) that can be possibly used as an input for the computeMatrix. Thus, I have used the (narrow Peaks) file from MACS2, after I converted it from a .tabular format into a. bed format, as an input for the computeMatrix tool. I then used the produced matrix as an input for the plotHeatmap tool in order to produce the desired heatmap that I wanted, and the whole thing seemed that it worked great.
Nevertheless, do you believe that the fact that I have used the (narrow Peaks) from MACS2 (after converted in .bed), as an input for the computeMatrix, was a good idea or not, concerning what will be finally depicted in the final Heatmap? I mean, all the computeMatrix is going to use are the three attributes from any provided .bed file that are referring to the chr, start and end of each peak, right? In that sense it should not matter, which of the files from MACS2 I would use, as long as it has these three attributes, am I correct? I would appreciate your input to this matter as well.